 |
What To Do When It's Not AMD
Learn how to identify and diagnose the multitude of other macular dystrophies and degenerations.
By Jessica Haynes, OD
Release Date: June 15, 2021
Expiration Date: June 15, 2024
Estimated Time to Complete Activity: 2 hours
Jointly provided by Postgraduate Institute for Medicine (PIM) and Review Education Group
Educational Objectives: After completing this activity, the participant should be better able to:
- Understand the pathophysiology of macular dystrophies and degenerations.
- Differentiate between common macular dystrophies and degenerations.
- Use the clinical exam techniques and tools to diagnose their patients.
- Understand how different macular conditions progress as well as their role in monitoring and managing care.
Target Audience: This activity is intended for optometrists engaged in eye care of macular dystrophies and degenerations.
Accreditation Statement: In support of improving patient care, this activity has been planned and implemented by the Postgraduate Institute for Medicine and Review Education Group. Postgraduate Institute for Medicine is jointly accredited by the Accreditation Council for Continuing Medical Education, the Accreditation Council for Pharmacy Education, and the American Nurses Credentialing Center, to provide continuing education for the healthcare team. Postgraduate Institute for Medicine is accredited by COPE to provide continuing education to optometrists.
Reviewed by: Salus University, Elkins Park, PA
Faculty/Editorial Board: Jessica Haynes, OD
Credit Statement: This course is COPE approved for 2 hours of CE credit. Activity #121904 and course ID 72958-PS. Check with your local state licensing board to see if this counts toward your CE requirement for relicensure.
Disclosure Statements:
Author: Dr. Haynes has no financial interests to disclose.
Managers and Editorial Staff: The PIM planners and managers have nothing to disclose. The Review Education Group planners, managers and editorial staff have nothing to disclose.
What Is A Dystrophy?This umbrella term loosely describes various progressive degenerative disorders. The term dystrophy also implies a monogenic or Mendelian inheritance, meaning the condition results from a specific variant of a single gene. Numerous degenerative conditions, such as AMD, are not considered dystrophies as they do not exhibit Mendelian inheritance. |
Optometrists have become well versed in the presentations of age-related macular degeneration (AMD), given its prevalence. Once that’s been firmly entrenched in your clinical skillset, to take it to the next level you’ll want to make sure that you’re inclusive of the non-AMD macular problems that present in your practice. By doing so, you’ll be able to isolate and differentially diagnose these conditions to better counsel and manage your patients.
The list of AMD masqueraders is lengthy and variable, including conditions that are degenerative, infectious, inflammatory, toxic, vascular, traumatic, neoplastic and paraneoplastic. Any condition that affects the retinal pigment epithelium (RPE) and outer retina may lead to drusenoid or lipofuscin deposition and/or pigmentary alteration that can mimic AMD.1 In this article, we will focus only on the non-AMD dystrophies and degenerations that affect the macula—a list that is already quite diverse and extensive.
What Stresses the Macula?
As it turns out, the lifelong responsibility of converting light energy into electrical potential to initiate the process of sight is a very stressful job. The photoreceptors, RPE and choroid must constantly work in sync to maintain the visual cycle, regenerate photoreceptor outer segments and remove and phagocytize metabolic waste products. Environmental factors such as UV light exposure and tobacco smoke put strain on this delicate balance, as do systemic conditions such as vascular disease. In addition, numerous faulty pathways can disrupt this system through various mechanisms.2,3
Phenotypical outcomes of various stressors can present similarly. Different pathways of damage may lead to clinically similar presentations that are difficult to distinguish from each other. Diagnostic imaging such as optical coherence tomography (OCT), fundus autofluorescence (FAF), fluorescein angiography (FA) and OCT angiography (OCT-A) alongside evaluation of retinal function with tools such as electrodiagnostics may help to narrow down a diagnosis. Additional factors such as age of onset, presenting symptoms and family history are also important, as these vary among different conditions.
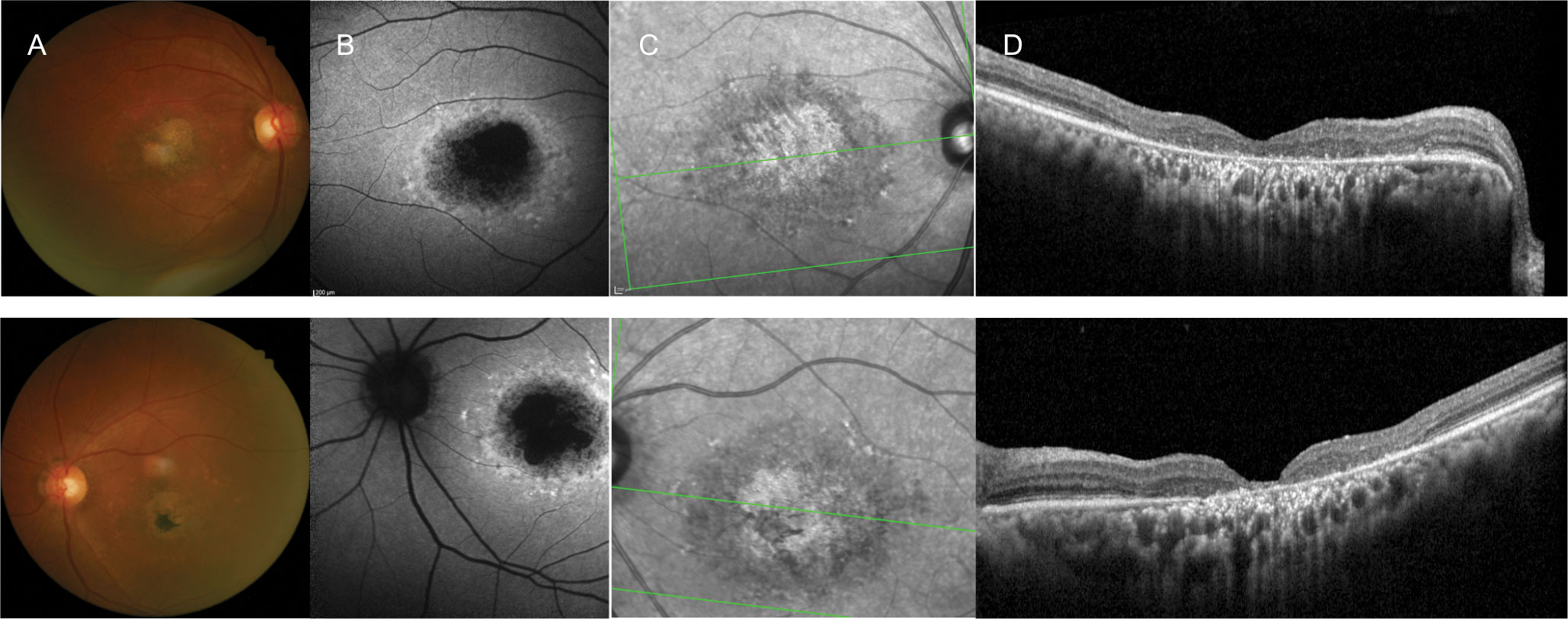 |
| Fig. 1. A 60-year-old white female with longstanding vision loss since her late teens and 20/150 best-corrected vision OD and OS. She presented with: (A) macular atrophy and surrounding yellow fleck lesions, (B) a bull’s eye-type pattern, (C) infrared reflectance OU on FAF and (D) outer retinal atrophy on OCT OU. One of her three siblings (a brother) had the same ocular condition. The others had normal vision, and there were no other affected family members, including her parents. The patient’s history was consistent with an autosomal recessively inherited condition, and she was clinically diagnosed with Stargardt’s disease. Genetic testing was offered, but she declined. Click image to enlarge. |
Stargardt’s Disease
The most commonly encountered inherited macular dystrophy, Stargardt’s, affects one in 8,000 to 10,000 individuals.4 Stargardt’s disease is most commonly inherited in an autosomal recessive fashion primarily by disease-causing variants of the ABCA4 gene.
This condition typically presents between the ages of 10 and 20, with a resultant visual acuity around 20/200.5,6 Presentation later in life usually results in better visual acuity outcomes. Patients often present with classic pisiform-shaped, yellow lesions or fleck-like lesions as well as macular atrophy with a “beaten bronze” appearance (Figure 1).6 Presentation of pisiform lesions without evidence of macular atrophy was initially termed fundus flavimaculatus, but is now recognized as a phenotypic variant of Stargardt’s disease.7
Diagnostic imaging is very useful in identifying and differentiating patients with Stargardt’s. Presentation is often subtle at first with visual symptoms being more severe than clinical signs.8 Care must be taken to identify these patients as to not misdiagnose them or perform unnecessary testing or procedures. OCT may show early thickening of the external limiting membrane. FAF may uncover early alterations and lipofuscin accumulation.8
In later stages, OCT imaging may demonstrate drusen-like subretinal, hyper-reflective deposits and varying amounts of photoreceptor atrophy and RPE disruption. FAF often reveals a reticular pattern of hyper-autofluorescent lipofuscin deposition. Areas of RPE atrophy will present as hypo-autofluorescent regions. In addition, a bull’s eye pattern of altered autofluorescence may be seen on FAF.9,10
The classic sign of Stargardt’s is a silent or dark choroid on FA with some citing its presence in up to 80% of patients.11 This sign is not present in all cases, however, and its absence cannot rule out Stargardt’s. While the condition is classified as an autosomal recessive condition, reports of altered visual function and retinal appearance have been described in carriers as well (Figure 2).12,13 Genetic testing should be considered to aid in diagnosis.
Electrodiagnostic testing is variable in patients with this condition. Pattern ERG and focal or multifocal ERG are typically significantly diminished or abolished, suggestive of macular disease. Some patients have a normal full-field ERG, while others have more widespread disease.14
There is currently no cure or treatment for Stargardt’s. Patients should be monitored for the rare occurrence of choroidal neovascularization (CNV). In addition, low vision training should be offered to those with reduced visual acuity. Confirming a genetic variant may be useful in light of clinical trials and future treatment options.
Phenotype vs. GenotypeThe attempt to discuss this group of conditions with strict, discrete categorization is very difficult due to their convoluted and often not entirely understood inheritance patterns alongside the phenotype/genotype conundrum. Genotype describes what genes are responsible for a particular condition, while phenotype outlines how a condition presents clinically. What does it actually look like? Even among family members who share the same genotype, the phenotypical presentation of the same condition may be inconsistent due to variable gene expression. Conditions were originally grouped primarily based on phenotypical appearance. With more information about these conditions and the genes that cause them, the classification and nomenclature used has evolved. However, this has left us with a bit of a mess to sift through when trying to give a name to a particular presentation. As genetic testing becomes more readily available and more is known about the genetic variants that cause certain conditions, we are able to arrive at more definitive clinical diagnoses. Access to genetic testing has significantly increased in recent years, becoming more standard of care in the management of inherited conditions. We must use the tools at our disposal along with patient demographics and our current knowledge and access to genetic testing to differentiate these conditions as best we can. |
Cone and Cone Rod Dystrophies
This heterogenous group of disorders involves progressive, widespread atrophy of cone photoreceptors leading to symptoms of visual acuity loss, decreased color vision and photophobia. In the same spectrum of disease are cone rod dystrophies that also involve rod photoreceptors.
Patients with clinically diagnosed cone dystrophies may eventually develop rod involvement with age leading to symptoms of nyctalopia and visual field loss. These conditions are genetically diverse and share affected genes with other retinal and macular dystrophies such as retinitis pigmentosa, considered a rod cone dystrophy, and Stargardt’s. Inheritance may be either autosomal dominant, recessive or x-linked.14-16
Clinical appearance, age of onset and visual outcome are variable. Visual acuity reduction typically presents in the first decade of life.14 Patients may present with pigmentary abnormalities, a bull’s eye macular appearance, macular atrophy or normal signs and symptoms.14-17 Those with cone rod dystrophy may later develop peripheral bone spicules.14 Varying levels of disc pallor are also reported.15
OCT is very useful in identifying photoreceptor atrophy, which may present as loss of the photoreceptor integrity line to more advanced loss of outer retinal tissue including the outer nuclear layer and RPE.14 FAF is useful in identifying alterations to the RPE that may not be readily visible clinically (Figure 3).14 The earliest finding on ERG is delayed 30Hz flicker implicit time followed by reduced 30Hz amplitude and reduced a-wave and b-wave amplitude with full-field photopic ERG. Those with cone rod dystrophies will later develop scotopic dysfunction.14 Genetic testing should be considered if available to aid in the diagnosis.
There is currently no cure or treatment for cone or cone rod dystrophies. Patients should be monitored for the rare occurrence of CNV. In addition, low vision training should be considered for those with impaired ability to perform activities associated with daily living. Identifying underlying genetic variants may be beneficial when considering clinical trials and future treatment options.
Pattern Dystrophies
This is an umbrella term that includes adult-onset vitelliform dystrophy (AOVD), butterfly-shaped pattern dystrophy (BSD), reticular dystrophy, multifocal pattern dystrophy simulating Stargardt’s and fundus pulverulentus. These conditions were initially categorized and classified based on clinical appearance. Pattern dystrophies in general were once thought to be inherited autosomal dominantly through disease-causing variants of the PRPH2 gene; however, a wide variety of affected genes and inheritance patterns are now being recognized.
In general, while pattern dystrophies are progressive conditions, patients tend to maintain good visual acuity. However, vision loss can occur from formation of macular atrophy, for which there is no treatment, or development of CNV, for which treatment with anti-VEGF is beneficial.18 Due to the development of visual symptoms later in life, these patients are more easily misdiagnosed with AMD.
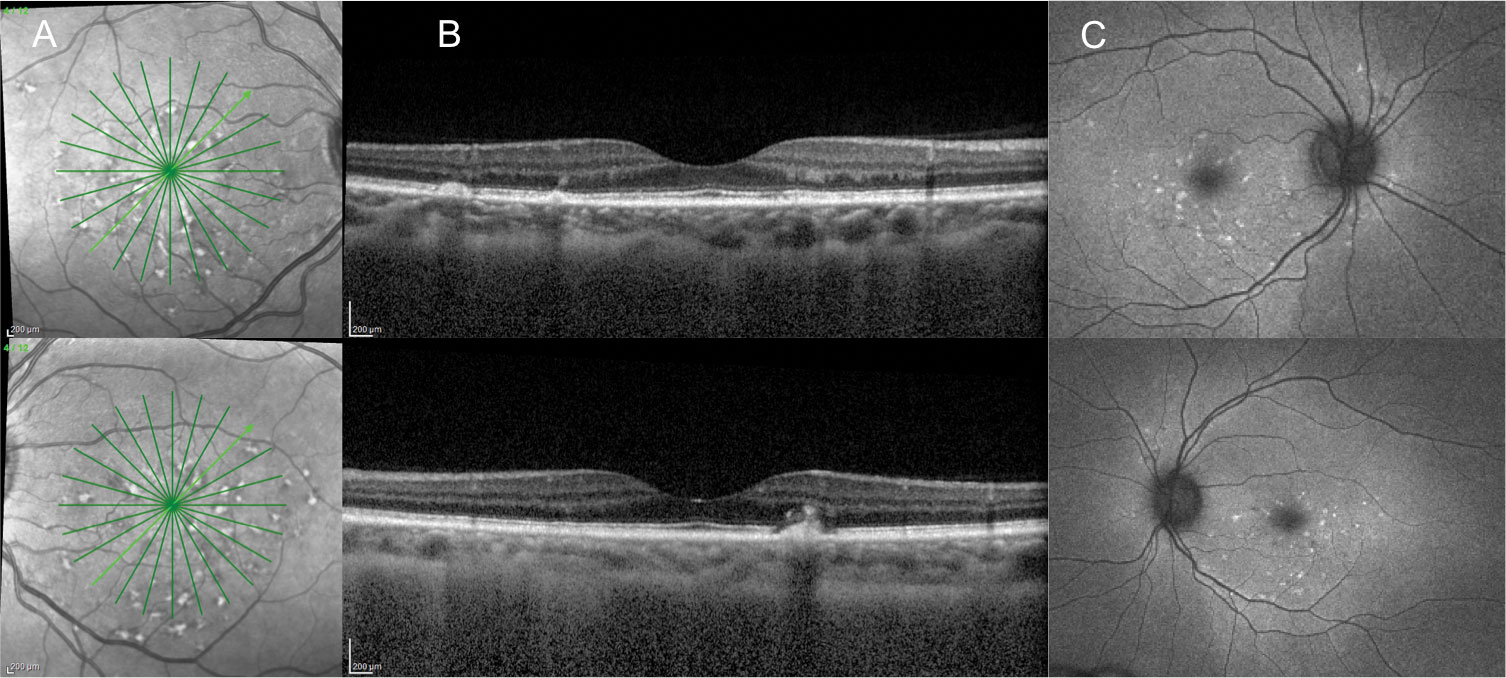 |
| Fig. 2. A 39-year-old Black female with 20/20 BCVA OD and OS presented with yellow, pisciform-shaped subretinal lesions. Images: (A) infrared photography reveals a reticular pattern of hyper-reflectance, (B) OCT shows areas of subretinal drusen-like deposits and RPE disruption and (C) FAF shows hyper-autofluorescence of the pisciform lesions. Genetic testing revealed she is an ABCA4 pathogenic variant carrier for Stargardt’s. Click image to enlarge. |
AOVD. This commonly encountered pattern dystrophy was first described as an autosomal dominant condition with bilateral, symmetric, circular subretinal lipofuscin deposits called vitelliform lesions (Figure 4).19 Since then, numerous publications have described the clinical, OCT, FAF and electrodiagnostic findings of the disease as well as its inheritance patterns. Confusion does exist in the literature due to the wide variety of names given to this condition as well as a lack of exact criteria needed to make the diagnosis.20 Another challenge is that vitelliform lesions can occur in a wide variety of outer retinal disease including AMD.
While the PRPH2 gene is causative in some patients with AOVD, a variety of other genes including BEST1 have been implicated in the condition as well. Most patients present without a family history, and in many, a responsible gene variant cannot be identified.20 This leads to the consideration that in some individuals AOVD may be more of a degenerative condition than a true dystrophy. Diagnosis is typically made in the sixth to eighth decade of life and is based on the clinical finding of bilateral, central vitelliform lesions.20 Variable amounts of additional RPE disruption and drusen deposition including reticular pseudodrusen have also been described.21
OCT is a useful diagnostic tool in this case. Vitelliform lesions present as hyper-reflective deposits between the RPE and the photoreceptors. Hypo-reflective regions of the lesion may also be present, causing confusion with the presence of “fluid.”22-24 On FAF, these lesions are typically hyper-autofluorescent since they are accumulations of lipofuscin (Figure 4).22,23 Electrodiagnostic testing is typically normal in these patients.20
BSD. The diagnosis of BSD is primarily clinical with a bilateral butterfly-shaped pattern of lipofuscin deposition and RPE disruption. OCT imaging reveals variable amounts of subretinal deposition and RPE disruption. FAF often highlights the butterfly-shaped pattern of disease. Patients are typically diagnosed in the second to third decade of life and usually have normal electrodiagnostic studies. The condition is still thought to be autosomal dominantly inherited due to variants in the PRPH2 gene.25,26
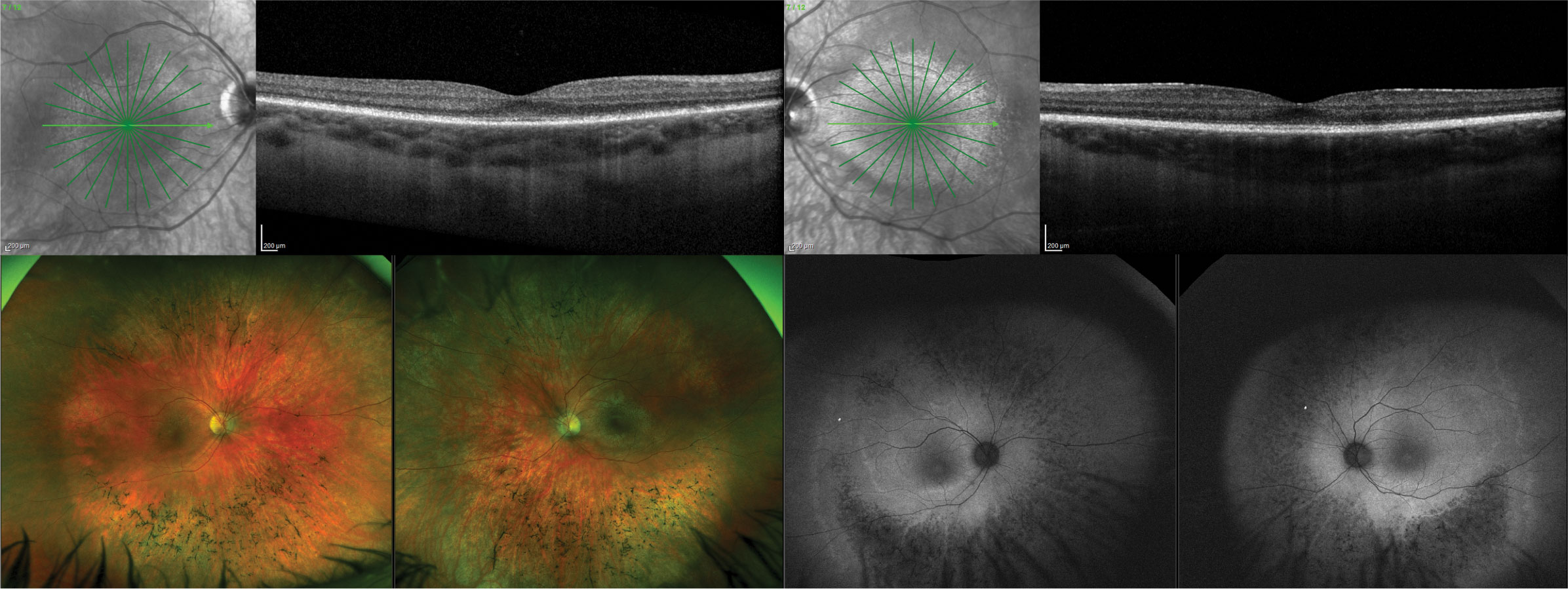 |
| Fig. 3. A 22-year-old white male presented with a homozygous pathogenic variant identified in TTLL5, consistent with cone rod dystrophy. OCT images show diffuse disruption of the photoreceptor integrity line. The macula has mild pigmentary alterations while the peripheral retina has a bone spicule-type appearance. FAF shows hypo-autofluorescence in the peripheral retina with hyper-autofluorescence centrally indicating both central (cone) and peripheral (rod) dysfunction. Click image to enlarge. |
Reticular dystrophy. This condition is typically diagnosed clinically by the presence of bilateral, subretinal yellow deposition and pigmentary alterations in a reticular pattern. OCT and FAF are also helpful in visualizing and distinguishing alterations to the outer retina and RPE in this disease as other pattern dystrophies.27
Multifocal pattern dystrophy simulating Stargardt’s. Considered to be inherited autosomal dominantly by pathogenic PRPH2 variants, this condition appears clinically and diagnostically similar to Stargardt’s disease (Figure 5). It may be differentiated through family history consistent with autosomal dominant inheritance; however, incomplete penetrance and variable expression may mask the inheritance pattern. Patients will typically present with findings later in life (fifth decade) and have on average a more stable disease course and better visual acuity later in life than those with Stargardt’s. In addition, they do not present with findings of a dark choroid on FA which is reported in the majority of Stargardt’s patients.28,29
Fundus pulverulentus. The least commonly encountered pattern dystrophy, fundus pulverulentus, is characterized by bilateral course pigment deposition in the macula. FA is helpful to show a typical pattern of hypo-fluorescent spots corresponding to the areas of pigment deposition. OCT and FAF findings have been rarely described.30-32
 |
| Fig. 4. A 69-year-old white female with AOVD has bilateral, circular, yellow, central vitelliform lesions (left). These lesions are hyper-autofluorescent on FAF (middle) and present on OCT as reflective subretinal deposits between the RPE and the photoreceptors (right). The patient presented with good visual acuity of 20/30 OD and OS. Click image to enlarge. |
Pseudoxanthoma Elasticum
PXE is caused by autosomal recessive inheritance of mutations on ABCC6, leading to calcification of elastic fibers in the eye, skin and vasculature. In the retina, this can lead to calcification of Bruch’s membrane causing pigmentary abnormalities such as peau d’orange and angioid streaks. Peau d’orange presents as a pebbly orange appearance of the retina typically in the temporal macula and mid-peripheral retina. Angioid streaks present as linear, radial pigmentary alterations extending from the optic nerve.33 In addition, PXE has been associated with pattern dystrophy-type appearances.32,34 PXE is a progressive condition in which vision loss can occur through tissue atrophy or development of CNV.35 Patients should be monitored carefully for development of CNV as it is common.
OCT can be used to image RPE and Bruch’s membrane alterations, and is also useful in identifying presence of CNV.33 Angioid streaks may be more apparent with FAF imaging than with fundus evaluation, and more diffuse alteration and atrophy of the RPE may be visible with FAF than on clinical examination.33,36 OCT-A and FA are useful for further evaluation and identification of CNV.37
Angioid streaks present during the lifetime of almost all patients with PXE, but they are not exclusive to PXE.38 They are also seen in patients with Ehlers-Danlos syndrome, Paget’s disease and sickle cell disease, and they may be idiopathic.
 |
| Click table to enlarge. |
Bestrophinopathies
This is a term used to describe a phenotypically heterogenous group of disorders caused by variants of the BEST1 gene. Most gene mutations lead to the phenotype consistent with Best disease, which we will focus on in this article.
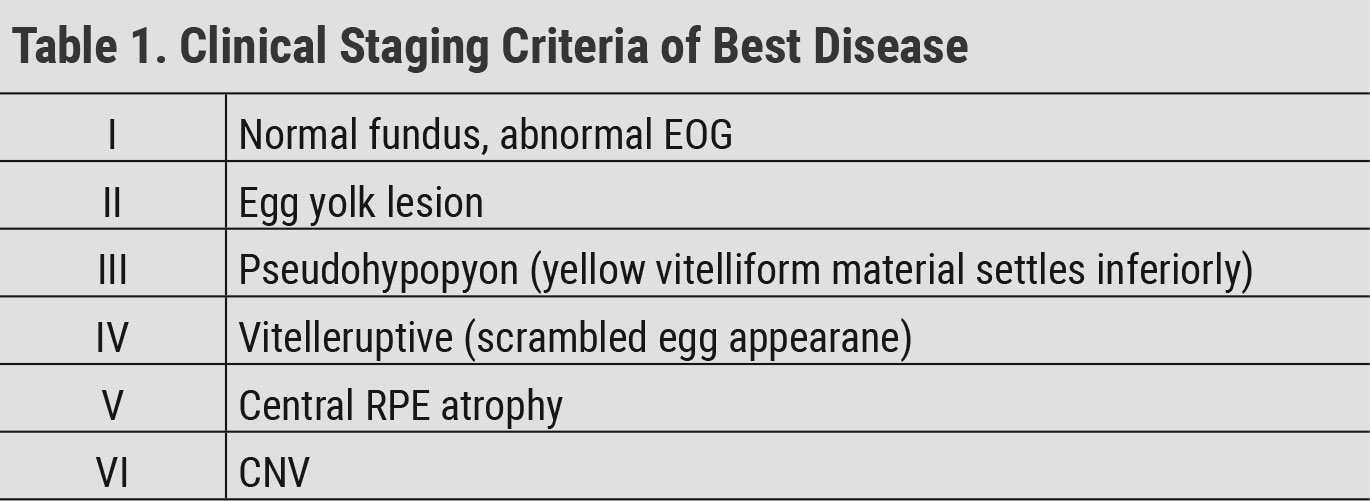
Best disease. An autosomal dominant dystrophy, this condition can present as early as the first decade of life. A generally accepted staging criteria for Best disease is shown in Table 1. Lesions in Best disease are typically bilateral and fairly symmetric. While lesions tend to be solitary, reports of multiple lesions per eye do exist. This has been termed “multifocal Best disease.”39 Patients often present with good visual acuity despite the striking fundus appearance. As the condition progresses, patients can develop loss of central acuity, metamorphopsias and central scotomas. Symptoms typically begin in the vitteleruptive stage with more severe loss occurring with progressive RPE atrophy or development of CNV. Sudden vision loss can occur with development of CNV.15
OCT is useful to image vitelliform lesions, which appear similarly to those in AOVD. On FAF, the lesions are typically hyper-autofluorescent due to the presence of lipofuscin. RPE atrophy may appear as hypo-autofluorescent. OCT-A and FA are useful in identifying the development of CNV.40 ERG is normal, but EOG is abnormal.15
 |
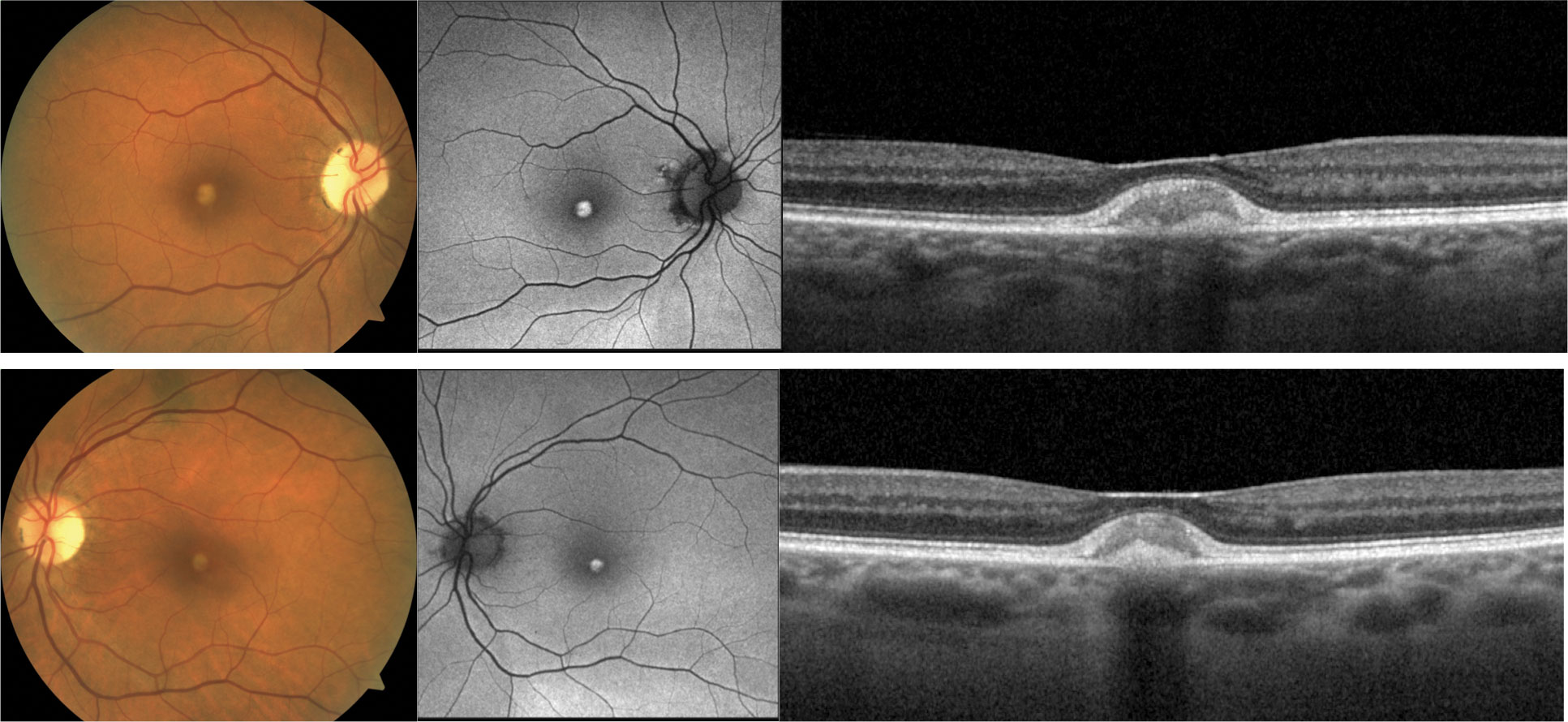
| Fig. 5. A 54-year-old white male presented with BCVA 20/70 OD and 20/50 OS. He complained of gradually worsening vision over the last two to three years. He had no family history of blindness. Fundus exam revealed bilateral yellow, fleck-type lesions scatted in the posterior pole and macular atrophy. FAF showed diffuse alteration of the autofluorescent signal. OCT showed hyper-reflective outer retinal deposits OU, photoreceptor atrophy OS and more extensive macular atrophy OD. The appearance is similar to Stargardt’s disease, but genetic testing identified a heterozygous pathogenic variant in PRPH2 more consistent with multifocal pattern dystrophy mimicking Stargardt’s. Click image to enlarge. |
Macular Drusen
Doyne honeycomb retinal dystrophy (DHRD) and Mallatia Leventinese (ML) are names both used to describe a phenotype of radially oriented macular drusen seen in relatively young individuals who may be in their 20s. These individuals often have peripapillary drusen as well.
The phenotype was first described by Walter Doyne in 1899 in Oxford, England, who coined the name DHRD.41 In 1932, a similar condition was described in several individuals in the Leventine Valley of Switzerland, then called ML.42 It is now generally accepted that these two names represent the same condition, caused by autosomal inheritance of a defect in the EFEMP1 gene that codes for a protein called fibulin 3, an extracellular matrix protein.43 Other names used to describe the phenotype are dominant drusen and familial drusen. However, evidence suggests that many patients with a phenotype of dominant drusen do not have the EFEMP1 gene mutation consistent with DHRD and ML.44,45
More recently, evaluation of drusen subtypes with multimodal imaging (OCT, FAF, intravenous FA, etc.) revealed that many in this young patient demographic have a phenotype of drusen called cuticular drusen. The term cuticular drusen was first used by Donald Gass in 1977 to describe drusen that appeared as numerous small hyper-fluorescent lesions on VA, appearing like a “starry sky.” This phenotype is currently being considered as a specific clinical subtype of AMD.46 Cuticular drusen seem to have a strong genetic component. Multiple genes are currently associated with the condition.18
OCT imaging can be used to visualize the drusen deposition and is also useful for the detection of CNV. Cuticular drusen often have a saw tooth-type appearance on OCT (Figure 6). FAF findings are variable, but FAF is useful to get a sense of the distribution and amount of RPE disruption and to identify regions of RPE atrophy.46 OCT-A and FA may be useful for identifying CNV. Electrophysiology testing in those confirmed with EFEMP1 macular disease has been reported as normal.47
Despite the significant amount of drusen seen on clinical examination, patients tend to present with good visual acuity. These conditions are progressive, however, with the possibility of visual decline from macular atrophy or development of CNV.46
 |
| Click table to enlarge. |
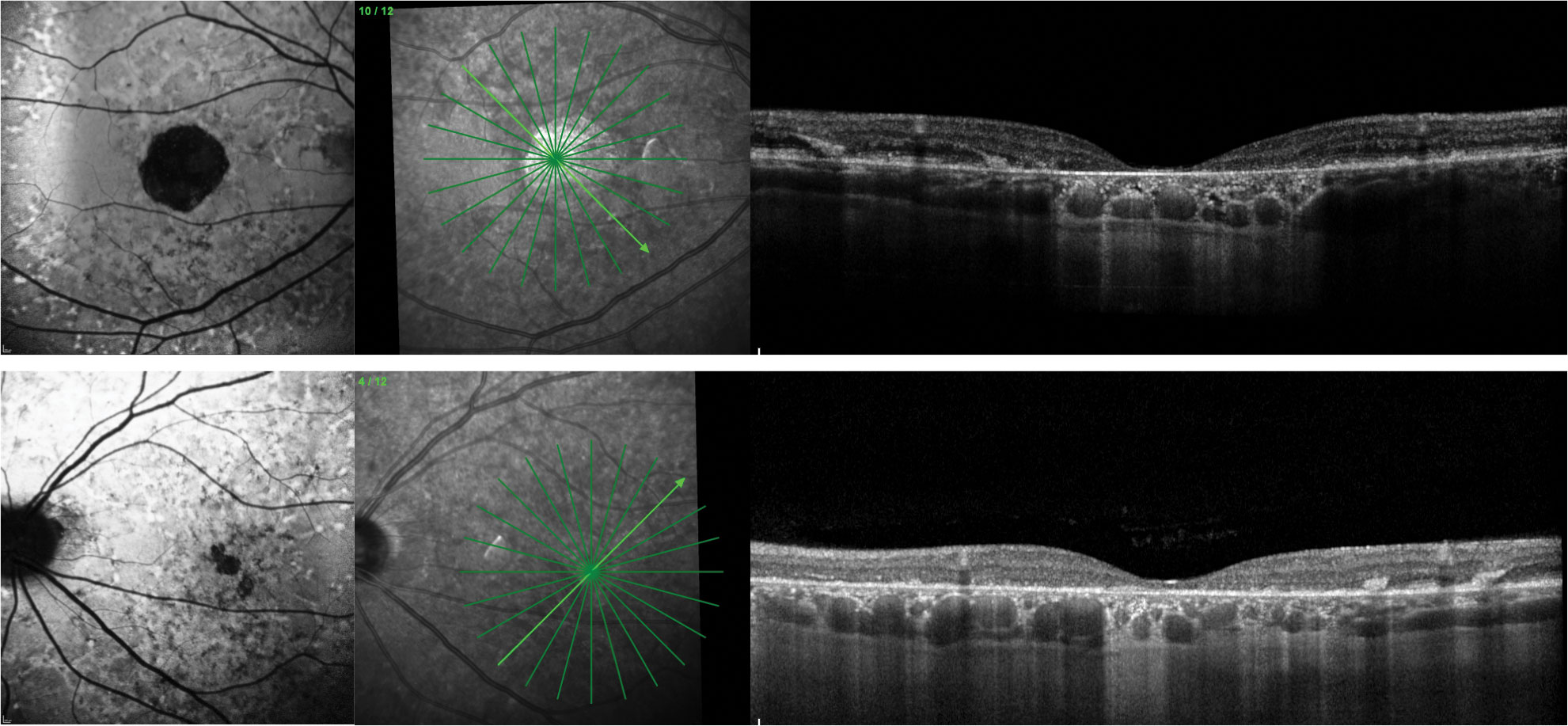
North Carolina Macular Dystrophy
This autosomal dominant condition has highly variable expressivity. A grading system has been created for the condition, but rather than acting as a staging criteria of disease progression, this is more of a staging of the phenotypic presentation as the condition is usually non-progressive (Table 2). The condition tends to be bilateral and symmetric. Vision loss is grade-dependent, but patients often have surprisingly good visual acuity on fundus presentation. The median acuity is reported to be 20/50. Patients of all grades must be monitored for development of CNV.15,48
Grade 3 lesions may appear excavated, and the terms staphyloma and coloboma have been used to describe them. Researchers describe this excavation as a deep chorioretinal excavation and have suggested the name “macular caldera” to describe these lesions.48 Dispute over the appropriate nomenclature for this excavation still exists.49-51
OCT and FAF can demonstrate the level of RPE and photoreceptor disruption. In addition, the deep chorioretinal excavation can be well visualized with OCT imaging. OCT-A and FA may be useful for identifying CNV. EOG and ERG are typically normal.15,48
 |
| Fig. 6. A 42-year-old Black male presented with a phenotypic appearance of autosomal dominant drusen. He had no family history and presented asymptomatically with 20/20 vision OD and OS. Images: (A) widefield photo, (B) FAF, (C) infrared reflectance and (D) OCT. Click image to enlarge. |
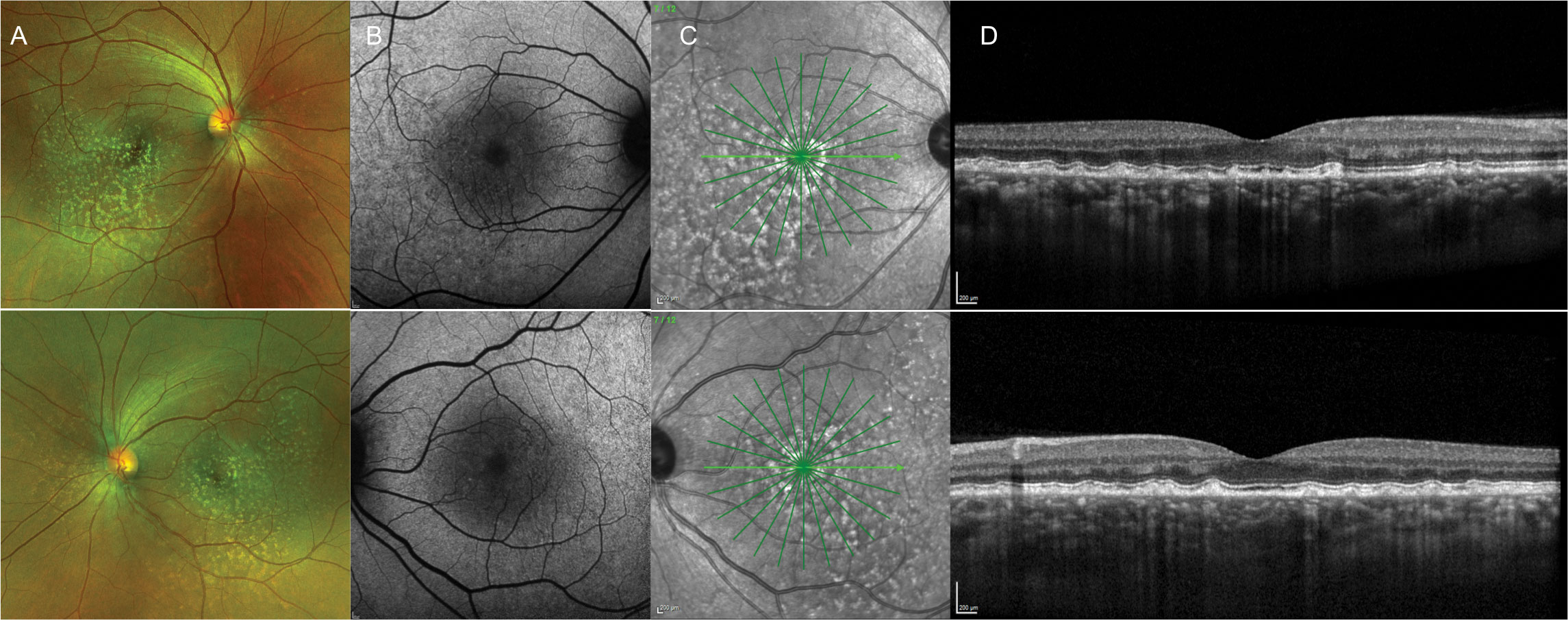
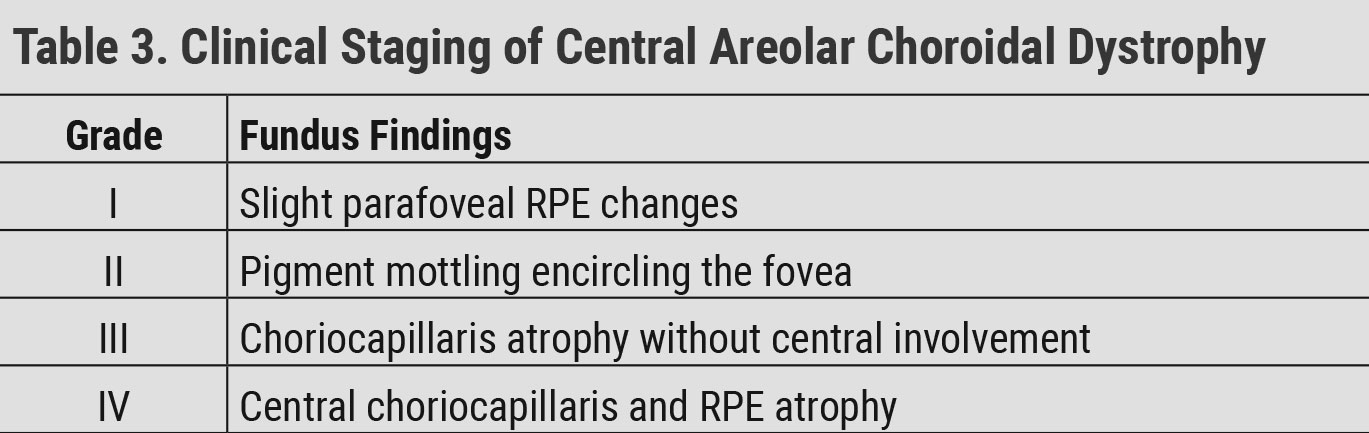
Central Areolar Choroidal Dystrophy
CACD is a rare autosomal dominant condition. A variant in PRPH2 is one known cause of CACD, but this is not the only gene identified in the condition. Patients present in the second decade of life with subtle bilateral, symmetrical pigment mottling centrally. This progresses to atrophy of the choriocapillaris, RPE and photoreceptors with vision loss beginning in the fourth and fifth decades of life.52 The condition presents as well defined, circular areas of macular atrophy. CACD progression has been staged in Table 3.15
Color vision is often abnormal, and later stages lead to visual acuity loss and central scotomas. Full-field ERG tends to be normal, aside from reduction in some advanced cases.15 Multifocal ERG has recently been reported to show dysfunction in broader regions than appear clinically diseased.53 Pattern VEP and pattern ERG are reported to be the earliest electrophysiological indicators of disease in patients with normal fundus appearance.53
OCT and FAF are very useful in detecting RPE disruptions from early to more advanced stages of CACD. Alterations may be subtle at first, but more advanced stages will show significant RPE and photoreceptor atrophy on OCT and FAF.54,55
 |
| Click table to enlarge. |
Myopic Macular Degeneration
MMD describes the atrophic changes that occur in highly myopic eyes, attributed to axial elongation. There is increased risk of MMD with higher refractive error, longer axial length, presence of posterior staphyloma involving the macula and older age (Figure 7). Findings in MMD include lacquer cracks (LCs), pigmentary alterations, macular atrophy and development of CNV.56
LCs are breaks in the RPE, Bruch’s membrane and choriocapillaris complex. These are typically seen in younger patients because as patients age, LCs often coalesce to form larger areas of macular atrophy.57 Patients with LCs can develop spontaneous, not CNV-related, subretinal hemorrhage due to choriocapillaris ruptures. This is a sign of likely LC expansion.58 In addition, MMD patients are at high risk for CNV development, and any presence of subretinal hemorrhage must be thoroughly investigated to rule out the presence of CNV on OCT, OCT-A and FA as needed.59
Macular atrophy can also develop without the presence of LCs. Regions of atrophy expand at variable rates with an increased risk of vision loss with age.56 While there is no treatment for macular atrophy, early detection of CNV and treatment with anti-VEGF can lead to better visual outcomes.60
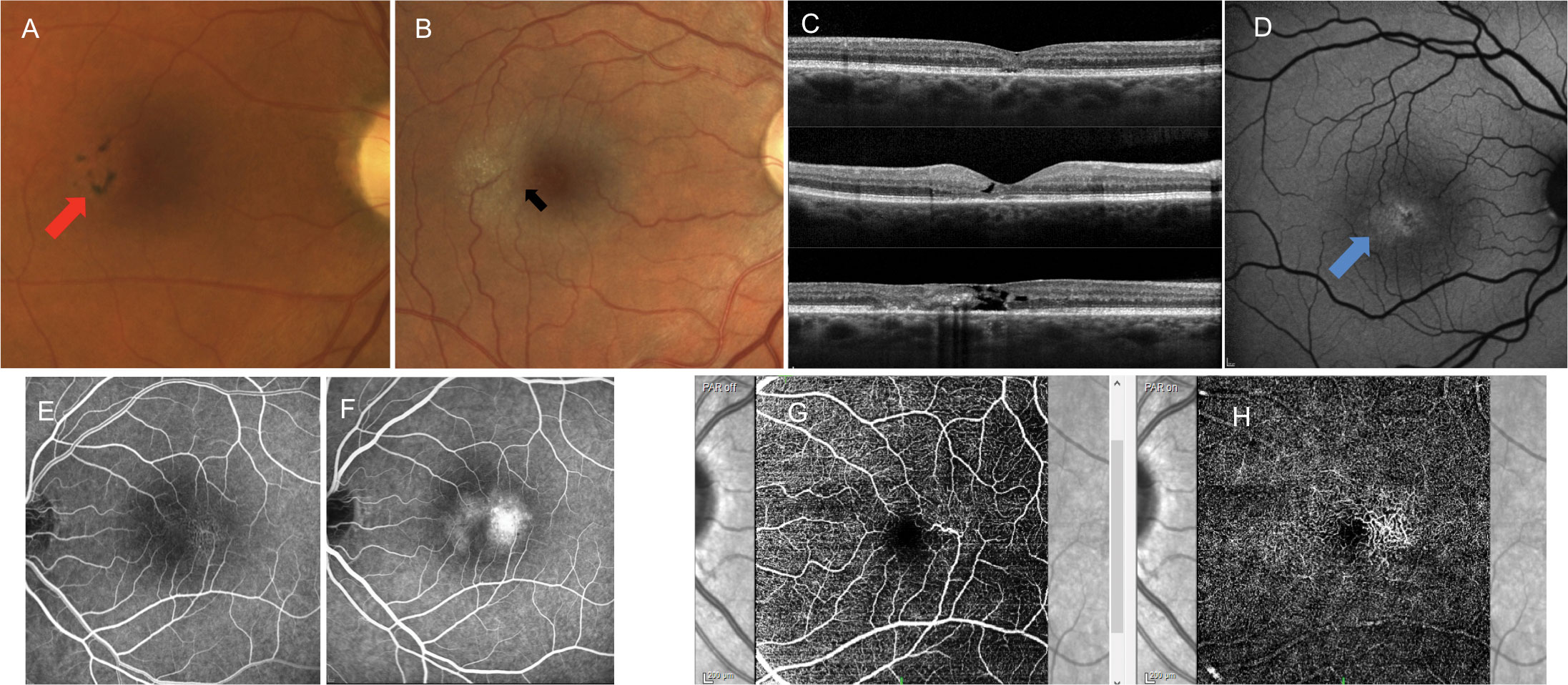 |
| Fig. 7. Patients with MT2 may present with the following findings: (A) pigmentary plaques (red arrow), (B) juxtafoveal whitening concentrated temporally, crystalline deposits and right angle vessels (black arrow), (C) variable retinal and photoreceptor atrophy and classic appearance of internal limiting membrane drape on OCT, (D) altered autofluorescent patterns typically showing first as hyper-autofluorescence temporally (blue arrow), (E) juxtafoveal telangiectatic vessels concentrated temporally on intravenous FA that leak in late stages and (F) telangiectatic vessels concentrated temporally in both the superficial (G) and deep (H) vascular plexus on OCT-A. These images are all examples of different patients. Click image to enlarge. |
Macular Telangiectasia Type 2
Patients with MT2 have abnormal parafoveal retinal capillaries most concentrated temporally. While these capillary abnormalities have given rise to the condition’s name, at its core MT2 is best described as a neurovascular macular degenerative condition. The pathophysiology is unclear, but it has been said that the cause may be from dysfunction in Muller cells that are vital for the maintenance of retinal health.61 While a genetic component is suspected, MT2 is not considered a macular dystrophy.61
Patients with MT2 tend to present with good visual acuity. The MacTel study group showed that 42% of all patients had best corrected vision of 20/25 or better.62 Symptoms tend to occur in the sixth or seventh decade of life with impaired reading being the most frequently reported initial symptom.63 Patients with MT2 may present with subtle retinal findings, making misdiagnosis easy (Figure 8). Initially, there is a mild, juxtafoveal retinal whitening most concentrated temporally. Later findings such as reflective deposits, pigmentary plaques and right-angle vessels may also be visible.61 The condition is bilateral, affecting the temporal juxtafoveal region to the greatest extent, but findings can be asymmetric.
The cause of vision loss in these patients stems from retinal and photoreceptor atrophy that typically affects the temporal juxtafoveal region. This can create scotomas in the presence of good central visual acuity, hence the difficulty with reading. Progression of the disease can lead to macular atrophy and decreased visual acuity. In addition, patients may develop CNV.61
OCT shows variable levels of retinal and photoreceptor atrophy. The presence of internal limiting membrane drape on OCT is classic for MT2. FAF shows hyper-autofluorescence temporally early in the disease with increased disruptions in the autofluorescent signal as the disease progresses. Pigmentary plaques appear as hypo-autofluorescent. Early-phase FA shows irregular juxtafoveal telangiectatic vessels most concentrated temporally. These vessels leak in the later stages, but the condition is not considered to be an exudative disease in the absence of CNV. This irregular vasculature can also be detected on OCT-A, showing evidence of telangiectatic vascular alterations most concentrated temporally in both the superficial and deep capillary plexus. OCT, FA and OCT-A are also helpful in identifying the presence of CNV.61
There is no treatment to slow the progression of MT2. Treatment of CNV with anti-VEGF has shown to be favorable.64
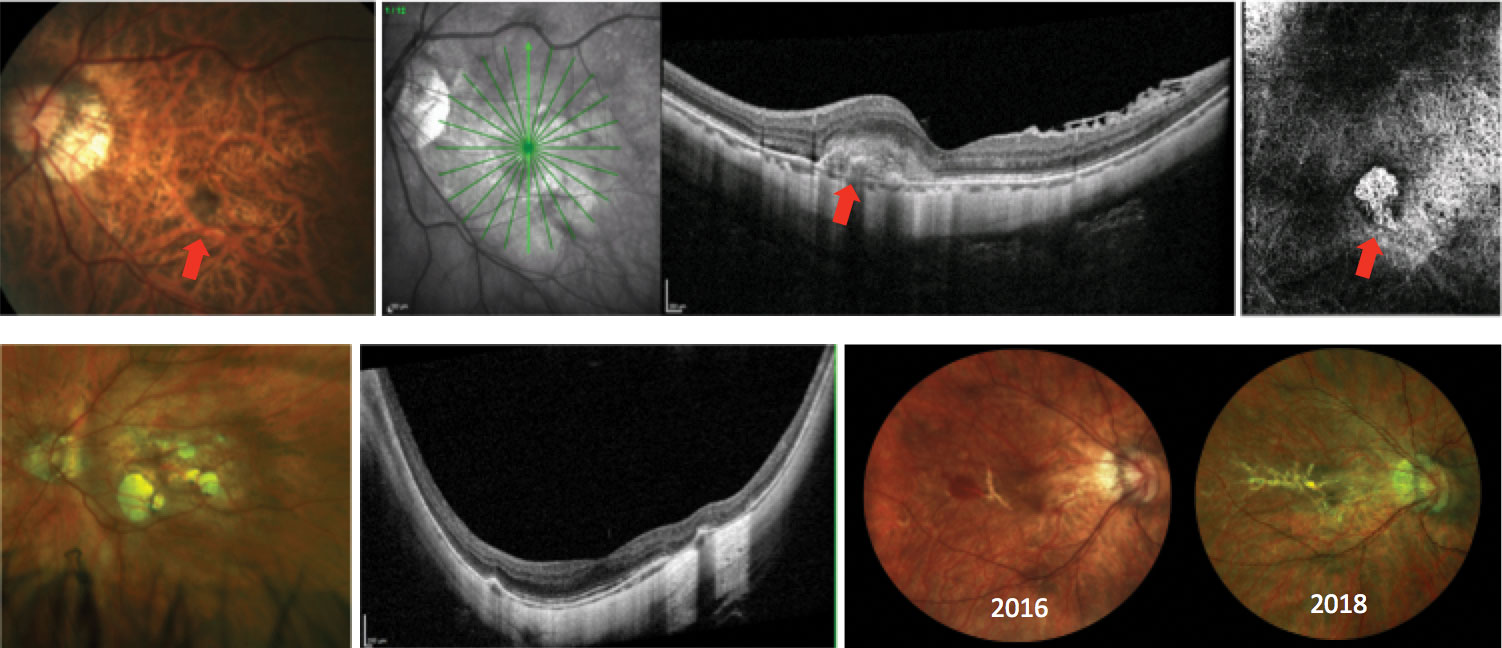 |
| Fig. 8. Various presentations of myopic degeneration. The top images show myopic CNV (red arrows) on fundus photo, OCT and in the avascular complex of the OCT-A. Bottom left shows a patient with posterior staphyloma and myopic macular atrophy. The bottom right shows progressive lacquer crack formation. In 2016, the patient had subretinal hemorrhage not associated with CNV. Click image to enlarge. |
Conclusions
When the macula encounters stress, either from extraneous sources or underlying defects in the system, the phenotypic results may not be unique to a particular condition. Findings such as drusen, lipofuscin deposition, pigmentary alterations, macular atrophy and CNV are seen in a wide variety of conditions.
Careful clinical examination along with imaging strategies such as OCT, FAF, OCT-A and FA help to guide us to a particular condition. Additional information such as electrodiagnostic studies, age of onset and family history also help to narrow the possibilities.
While we are extremely limited in our treatment options for macular dystrophies and degenerations, arriving at an accurate diagnosis allows us to educate patients and their family members about their visual prognoses. It can also help guide decisions on the value of genetic testing.
All patients with macular degenerations or dystrophies are at increased risk of developing CNV and should be monitored for this occurrence. In general, treatment with intravitreal anti-VEGF is favorable in those who develop CNV. Those left with visual impairments that affect their daily routine should be referred to a low vision specialist.
Dr. Haynes is a consultative optometrist at the Charles Retina Institute in Germantown, TN, and a consulting faculty member at the Southern College of Optometry in Memphis, TN. She has no financial interests to disclose.
| 1. Paez-Escamilla M, Jhingan M, Gallagher DS, et al. Age-related macular degeneration masqueraders: from the obvious to the obscure. Surv Ophthalmol. 2021;66(2):153-82. 2. Tsin A, Betts-Obregon B, Grigsby J. Visual cycle proteins: structure, function, and roles in human retinal disease. J Biol Chem. 2018;293(34):13016-21. 3. Bird A. Role of retinal pigment epithelium in age-related macular disease: a systematic review. Br J Ophthalmol. September 19, 2020. [Epub ahead of print]. 4. Georgiou M, Kane T, Tanna P, et al. Prospective cohort study of childhood-onset Stargardt disease: fundus autofluorescence imaging, progression, comparison with adult-onset disease, and disease symmetry. Am J Ophthalmol. 2020;211:159-75. 5. Walia S, Fishman GA. Natural history of phenotypic changes in Stargardt macular dystrophy. Ophthalmic Genet. 2009;30(2):63-8. 6. Valkenburg D, Runhart EH, Bax NM, et al. Highly variable disease courses in siblings with Stargardt disease. Ophthalmology. 2019;126(12):1712-21. 7. Haji Abdollahi S, Hirose T. Stargardt-fundus flavimaculatus: recent advancements and treatment. Semin Ophthalmol. 2013;28(5-6):372-6. 8. Bax NM, Lambertus S, Cremers FPM, et al. The absence of fundus abnormalities in Stargardt disease. Graefes Arch Clin Exp Ophthalmol. 2019;257(6):1147-57. 9. Arrigo A, Grazioli A, Romano F, et al. Multimodal evaluation of central and peripheral alterations in Stargardt disease: a pilot study. Br J Ophthalmol. 2020;104(9):1234-8. 10. Chen Y, Roorda A, Duncan JL. Advances in imaging of Stargardt disease. Adv Exp Med Biol. 2010;664:333-40. 11. Fishman GA, Farber M, Patel BS, et al. Visual acuity loss in patents with Stargardt’s macular dystrophy. Ophthalmology. 1987;94(7):809-14. 12. Kjellström U. Reduced macular function in ABCA4 carriers. Mol Vis. 2015;21:767-82. 13. Duncker T, Stein GE, Lee W, et al. Quantitative fundus autofluorescence and optical coherence tomography in ABCA4 carriers. Invest Ophthalmol Vis Sci. 2015;56(12):7274-85. 14. Hoyt C, Taylor D. Pediatric Ophthalmology and Strabismus. 4th ed., Saunders/Elsevier, 2012. 15. Gill JS, Georgiou M, Kalitzeos A, et al. Progressive cone and cone-rod dystrophies: clinical features, molecular genetics and prospects for therapy. Br J Ophthalmol. 2019;103(5):711-20. 16. Hamel CP. Cone rod dystrophies. Orphanet J Rare Dis. 2007;2(1):7. 17. Kominami A, Ueno S, Kominami T, et al. Case of cone dystrophy with normal fundus appearance associated with biallelic POC1B variants. Ophthalmic Genet. 2018;39(2):255-62. 18. Duvvari MR, van de Ven JPH, Geerlings MJ, et al. Whole exome sequencing in patients with the cuticular drusen subtype of age-related macular degeneration. PLoS One. 2016;11(3):e0152047. 19. Gass JD. A clinicopathologic study of a peculiar foveomacular dystrophy. Trans Am Ophthalmol Soc. 1974;72:139-56. 20. Chowers I, Tiosano L, Audo I, et al. Adult-onset foveomacular vitelliform dystrophy: a fresh perspective. Prog Retin Eye Res. 2015;47:64-85. 21. Wilde C, Lakshmanan A, Patel M, et al. Prevalence of reticular pseudodrusen in newly presenting adult onset foveomacular vitelliform dystrophy. Eye (Lond). 2016;30(6):817-24. 22. Grob S, Yonekawa Y, Eliott D. Multimodal imaging of adult-onset foveomacular vitelliform dystrophy. Saudi J Ophthalmol. 2014;28(2):104-10. 23. Bastos RR, Ferreira CS, Brandão E, et al. Multimodal image analysis in acquired vitelliform lesions and adult-onset foveomacular vitelliform dystrophy. J Ophthalmol. 2016;2016:6037537. 24. Regatieri CV, Branchini L, Duker JS. The role of spectral-domain OCT in the diagnosis and management of neovascular age-related macular degeneration. Ophthalmic Surg Lasers Imaging. 2011;42:S56-66. 25. Zhang K, Garibaldi DC, Li Y, et al. Butterfly-shaped pattern dystrophy: a genetic, clinical, and histopathological report. Arch Ophthalmol. 2002;120(4):485-90. 26. Kumar V, Kumawat D. Multimodal imaging in a case of butterfly pattern dystrophy of retinal pigment epithelium. Int Ophthalmol. 2018;38(2):775-9. 27. Zerbib J, Querques G, Massamba N, et al. Reticular pattern dystrophy of the retina: a spectral-domain optical coherence tomography analysis. Am J Ophthalmol. 2013;156(6):1228-37. 28. Boon CJF, Van Schooneveld MJ, Den Hollander AI, et al. Mutations in the peripherin/RDS gene are an important cause of multifocal pattern dystrophy simulating STGD1/fundus flavimaculatus. Br J Ophthalmol. 2007;91(11):1504-11. 29. Roy R, Kumar S, Chandrasekharan DP, et al. Multimodal imaging in multifocal pattern dystrophy simulating fundus flavimaculatus. Indian J Ophthalmol. 2016;64(5):395-6. 30. Parodi MB. Choroidal neovascularization in fundus pulverulentus. Acta Ophthalmol Scand. 2002;80(5):559-60. 31. Roy R, Saurabh K, Shah D. Multimodal imaging in a case of fundus pulverulentus. Retina. 2018;38(7):e55-8. 32. Ebran JM, Martin L, Leftheriotis, et al. Subretinal fibrosis is associated with fundus pulverulentus in pseudoxanthoma elasticum. Graefes Arch Clin Exp Ophthalmol. 2018;256(4):699-707. 33. Gliem M, De Zaeytijd J, Finger RP, et al. An update on the ocular phenotype in patients with pseudoxanthoma elasticum. Front Genet. 2013;4:14. 34. Agarwal A, Patel P, Adkins T, et al. Spectrum of pattern dystrophy in pseudoxanthoma elasticum. Arch Ophthalmol. 2005;123(7):923-8. 35. Finger RP, Issa CP, Ladewig M, et al. Intravitreal bevacizumab for choroidal neovascularisation associated with pseudoxanthoma elasticum. Br J Ophthalmol. 2008;92(4):483-7. 36. Shiraki K, Kohno T, Moriwaki M, et al. Fundus autofluorescence in patients with pseudoxanthoma elasticum. Int Ophthalmol. 2001;24(5):243-8. 37. Birtel J, Lindner M, Mishra DK, et al. Retinal imaging including optical coherence tomography angiography for detecting active choroidal neovascularization in pseudoxanthoma elasticum. Clin Exp Ophthalmol. 2019;47(2):240-9. 38. Orssaud C, Roche O, Dufier JL, et al. Visual impairment in pseudoxanthoma elasticum: a survey of 40 patients. Ophthalmic Genet. 2015;36(4):327-32. 39. Boon CJF, Klevering BJ, Leroy BP, et al. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res. 2009;28(3):187-205. 40. Ferrara DC, Costa RA, Tsang S, et al. Multimodal fundus imaging in Best vitelliform macular dystrophy. Graefes Arch Clin Exp Ophthalmol. 2010;248(10):1377-86. 41. Doyne RW. Peculiar condition of choroiditis occurring in several members of the same family. Trans Ophthalmol Soc UK. 1899;19:71. 42. Klainguti R. Die tapetoretinal degeneration im kanton tessin. Klin Monatsbl Augenheilkd. 1932;89:253-4. 43. Stone EM, Lotery AJ, Munier FL, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999;22(2):199-202. 44. Tarttelin EE, Gregory-Evans CY, Bird AC, et al. Molecular genetic heterogeneity in autosomal dominant drusen. J Med Genet. 2001;38(6):381-4. 45. Sauer CG, White K, Kellner U, et al. EFEMP1 is not associated with sporadic early onset drusen. Ophthalmic Genet. 2001;22(1):27-34. 46. Balaratnasingam C, Cherepanoff S, Dolz-Marco R, et al. Cuticular drusen: clinical phenotypes and natural history defined using multimodal imaging. Ophthalmology. 2018;125(1):100-18. 47. Haimovici R, Wroblewski J, Piguet B, et al. Symptomatic abnormalities of dark adaptation in patients with EFEMP1 retinal dystrophy (Malattia Leventinese/Doyne honeycomb retinal dystrophy). Eye (Lond). 2002;16(1):7-15. 48. Khurana RN, Sun X, Pearson E, et al. A reappraisal of the clinical spectrum of North Carolina macular dystrophy. Ophthalmology. 2009;116(10):1976-83. 49. Schoenberger SD, Agarwal A. Intrachoroidal cavitation in North Carolina macular dystrophy. JAMA Ophthalmol. 2013;131(8):1073-6. 50. Agarwal A, Schoenberger SD. Macular caldera in North Carolina macular dystrophy: only an illusion of posterior pole staphyloma-reply. JAMA Ophthalmol. 2014;132(6):787. 51. Chen C, Khurana RN, Scholl H, et al. Macular caldera in North Carolina macular dystrophy: only an illusion of posterior pole staphyloma. JAMA Ophthalmol. 2014;132(6):786-7. 52. Gundogan FC, Dinç UA, Erdem U, et al. Multifocal electroretinogram and central visual field testing in central areolar choroidal dystrophy. Eur J Ophthalmol. 2010;20(5):919-24. 53. Lotery AJ, Silvestri G, Collins AD. Electrophysiology findings in a large family with central areolar choroidal dystrophy. Doc Ophthalmol. 1998;97(2):103-19. 54. Boon CJF, Klevering BJ, Cremers FPM, et al. Central areolar choroidal dystrophy. Ophthalmology. 2009;116(4):771-82. 55. Smailhodzic D, Fleckenstein M, Theelen T, et al. Central areolar choroidal dystrophy (CACD) and age-related macular degeneration (AMD): differentiating characteristics in multimodal imaging. Invest Ophthalmol Vis Sci. 2011;52(12):8908-18. 56. Ruiz-Medrano J, Montero JA, Flores-Moreno I, et al. Myopic maculopathy: current status and proposal for a new classification and grading system (ATN). Prog Retin Eye Res. 2019;69:80-115. 57. Ohno-Matsui K, Tokoro T. The progression of lacquer cracks in pathologic myopia. Retina. 1996;16(1):29-37. 58. Ohno-Matsui K, Ito M, Tokoro T. Subretinal bleeding without choroidal neovascularization in pathologic myopia. A sign of new lacquer crack formation. Retina. 1996;16(3):196-202. 59. Mi L, Zuo C, Zhang X, et al. Fluorescein leakage within recent subretinal hemorrhage in pathologic myopia: suggestive of CNV? J Ophthalmol. 2018;2018:4707832. 60. Cheung CMG, Arnold JJ, Holz FG, et al. Myopic choroidal neovascularization. Ophthalmology. 2017;124(11):1690-1711. 61. Issa PC, Gillies MC, Chew EY, et al. Macular telangiectasia type 2. Prog Retin Eye Res. 2013;34:49-77. 62. Clemons TE, Gillies MC, Chew EY, et al. Baseline characteristics of participants in the natural history study of macular telangiectasia (MacTel) MacTel Project Report No. 2. Ophthalmic Epidemiology. 2010:17(1):66-73. 63. Heeren TFC, Holz FG, Issa PC. First symptoms and their age of onset in macular telangiectasia type 2. Retina. 2014;34(5):916-9. 64. Khodabande A, Roohipoor R, Zamani J, et al. Management of idiopathic macular telangiectasia type 2. Ophthalmol Ther. 2019;8(2):155-75. |