Unlocking the Mystery of Corneal Dystrophies
Optometrists are often the first to detect the clues and sleuth out the diagnosis. Here’s what to look for.
By Mitch Ibach, OD, and Larae Zimprich, OD
 |
Release Date: November 15, 2019
Expiration Date: November 15, 2022
Estimated time to complete activity: 1 hour
Jointly provided by Postgraduate Institute for Medicine and Review Education Group.
Educational Objectives: After completing this activity, the participant should be better able to:
- Identify the most common traits as well as the characteristic signs and symptoms related to corneal dystrophies.
- Consider the differential diagnosis for corneal dystrophies.
- Perform the necessary elements of the patient history, ocular examination/vision testing and laboratory testing required to diagnose corneal dystrophies.
- Distinguish the different types of corneal dystrophies most commonly encountered by optometrists, with at least a cursory understanding of their genetic inheritance.
- Provide, or otherwise obtain, the ocular and systemic treatment that the patient requires.
Target Audience: This activity is intended for optometrists engaged in the care of patients with corneal dystrophies.
Accreditation Statement: In support of improving patient care, this activity has been planned and implemented by the Postgraduate Institute for Medicine and Review Education Group. Postgraduate Institute for Medicine is jointly accredited by the Accreditation Council for Continuing Medical Education, the Accreditation Council for Pharmacy Education, and the American Nurses Credentialing Center, to provide continuing education for the healthcare team. Postgraduate Institute for Medicine is accredited by COPE to provide continuing education to optometrists.
Faculty/Editorial Board: Mitch Ibach, OD, Vance Thompson Vision, Sioux Falls, SD, and Larae Zimprich, OD, Vance Thompson Vision.
Credit Statement: This course is COPE approved for 1 hour of CE credit. Course ID is 65003-AS. Check with your local state licensing board to see if this counts toward your CE requirement for relicensure.
Disclosure Statements:
Dr. Ibach receives consulting fees from Aerie, Alcon, Avedro, Glaukos, Ocular Therapeutix and fees for non-CME/CE services from Aerie and Glaukos; and has ownership interest in Equinox.
Dr. Zimprich has no disclosures.
Managers and Editorial Staff: The PIM planners and managers have nothing to disclose. The Review Education Group planners, managers and editorial staff have nothing to disclose.
Corneal dystrophies are a group of progressive inheritable conditions that, over time, cause bilateral pathology. The manifestations, which are commonly deposits of material in the anterior cornea and cell atrophy in the posterior cornea, are not associated with trauma, infection or inflammation; rather, they are secondary to genetic mutations.1
Although corneal dystrophy is a rare condition, the familial impact makes diagnosis and appropriate counseling of the utmost importance.
This review aims to unlock the mystery of corneal dystrophies by discussing prevalence, genetic patterns, slit lamp clues, treatments and other pertinent points to help clinicians better diagnose and care for these patients.
Classification of corneal dystrophies is routinely based on the affected corneal layer. A recent epidemiologic retrospective analysis of managed care patients revealed a prevalence of a single corneal dystrophy at 0.13%.1 Endothelial dystrophies were the most common (60.4% of the total) followed by anterior dystrophies (15.6%). Lastly, stromal dystrophies were identified as the least frequent.1 However, 26% of cases in this study were listed as “unspecified” dystrophies.
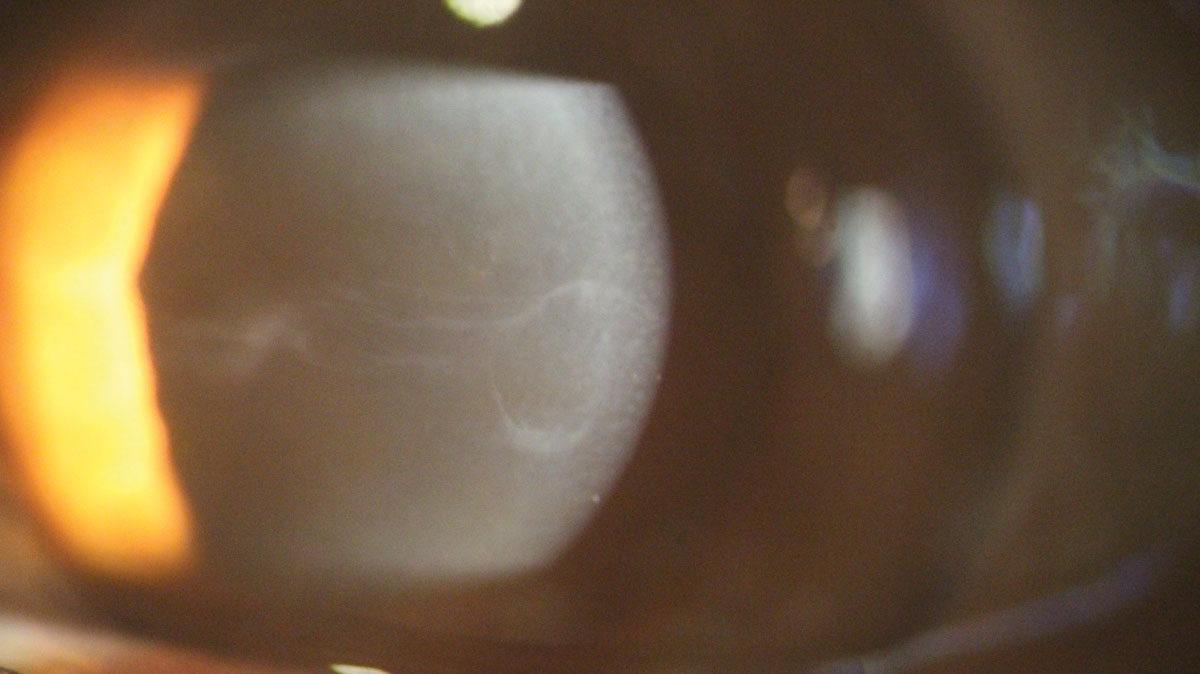 |
| Central epithelial basement membrane dystrophy on high magnification. Click image to enlarge. |
Anterior Corneal Dystrophies
Four main corneal dystrophies affect the superficial cornea: epithelial basement membrane dystrophy (EBMD), Meesmann dystrophy, Reis-Bucklers dystrophy and Thiel-Behnke dystrophy. The epithelium, approximately 50μm thick, serves as a barrier to provide protection and allow the diffusion of oxygen from the tear film. All of these dystrophies can compromise the epithelium, which often leads to pain and irregular astigmatism, which causes poor visual outcomes.
Epithelial basement membrane dystrophy. EBMD is a bilateral, though often asymmetric, condition characterized by thickening and redundancy of the corneal epithelial basement membrane and the abnormal adhesion to the basal epithelial cells. It is not uncommon for patients to be asymptomatic or even unaware they have the condition.
There are reported cases of EBMD with studied point mutations in the TGFβI gene on chromosome 5 in an autosomal dominant pattern; however, some feel that it is more of a degeneration rather than a true dystrophy.2 The genetic inheritance pattern is rather weak, which leads some people to believe it is a degeneration associated with aging, dry eye and trauma.
Patients who are symptomatic may complain of blurred vision due to induced irregular astigmatism, mild to severe pain, sensitivity to light, tearing or a gritty, sandy-like feeling. Oftentimes, a patient’s symptoms can be exacerbated upon awakening due to the detachment of damaged epithelium, which may cause significant pain. Once this occurs, the cornea may become prone to recurrent corneal erosion (RCE). Approximately 10% of patients with EBMD experience recurrent corneal erosions and, conversely, 50% of patients with RCE evidence EBMD in the contralateral eye.3
Although EBMD is one of the most common corneal dystrophies, it may present in a variety of ways. Slit-lamp examination of a patient with EBMD may reveal bilateral subepithelial microcysts, chalky patches, thickened gray areas that resemble fingerprints or map-like lesions—giving us the term “map-dot-fingerprint” dystrophy. These findings are confined to the epithelial layer, and negative staining in a whorl-like pattern can be observed with the instillation of fluorescein.
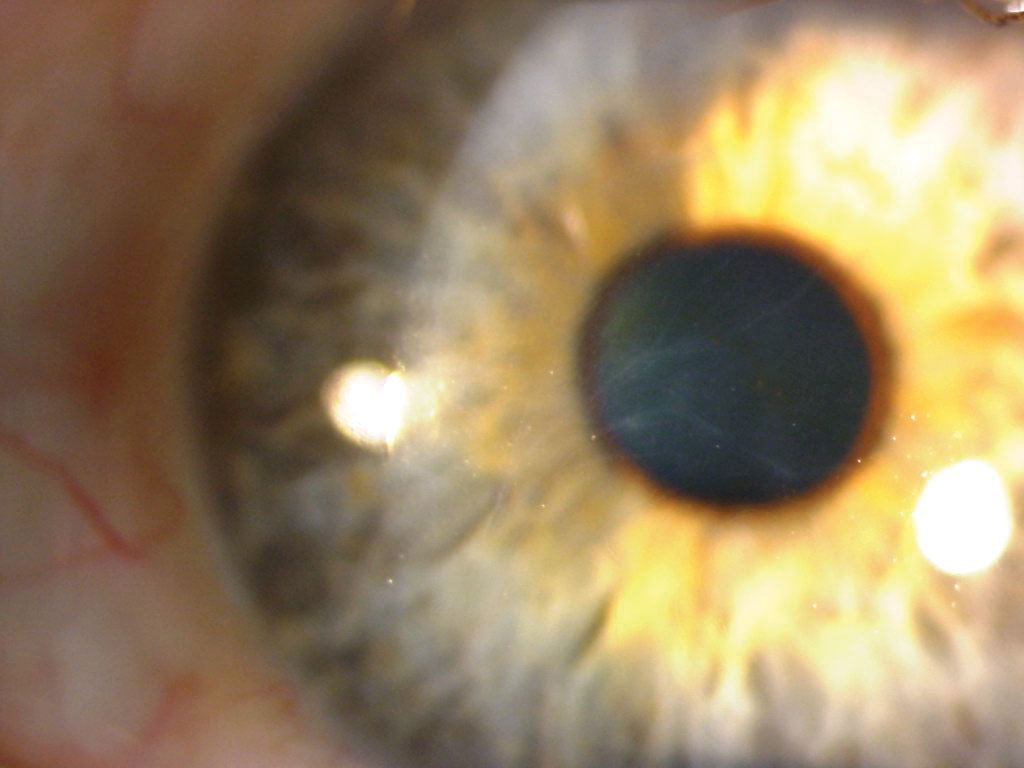 |
EBMD in a patient with a history of fingernail trauma. Click image to enlarge. |
Meesmann dystrophy. This is an autosomal-dominant epithelial corneal dystrophy. Meesmann dystrophy typically manifests from a young age and is caused by a defect in the KRTI2 gene, which leads to thickening of the epithelial basement membrane, similarly to EBMD.4 However, slit-lamp examination reveals bilateral, small, punctate or bubble-like intraepithelial cysts concentrated in the interpalpebral region.
Reis-Bucklers and Thiel-Behnke dystrophies. These are Bowman’s layer dystrophies caused by mutations in TGFβI that manifest later in life compared with Meesmann corneal dystrophy.4 Corneal examination in a patient with Reis-Bucklers or Thiel-Behnke dystrophy may reveal subepithelial deposits of hyaline-like material that damage and eventually replace Bowman’s layer.2 The cornea may also show dense gray-white subepithelial opacities centrally that worsen with age.
Although clinically similar, they can be differentiated from one another by the characteristic finding of curled filaments within Bowman’s membrane on confocal microscopy, which is pathognomonic for Thiel-Behnke.4
RCEs may occur in all epithelial and Bowman’s dystrophies due to ruptured cysts and an unstable epithelium; however, they are more common in Reis-Bucklers and Thiel-Behnke dystrophies.4 Otherwise, typical symptoms such as glare, light sensitivity and blurred vision are generally mild.
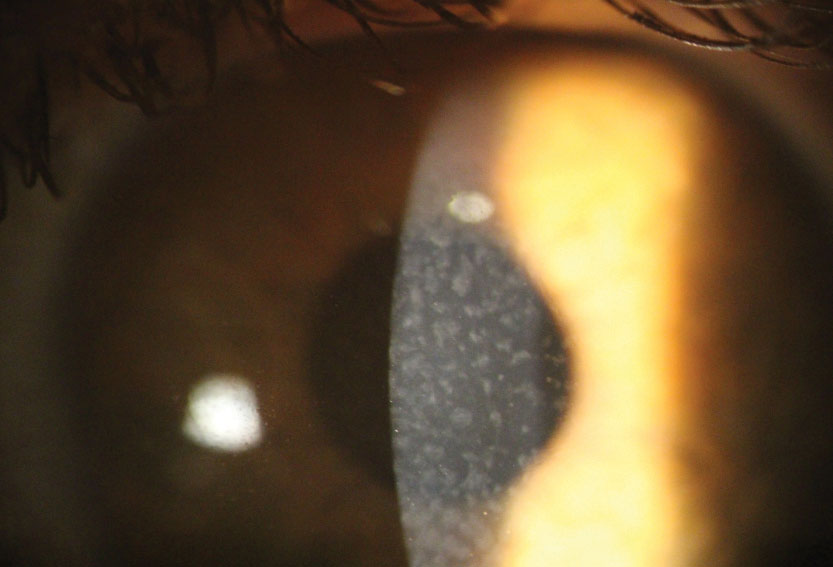 |
| Reis-Bucklers dystrophy with central grayish opacities in Bowman’s layer. Click image to enlarge. |
Treatment of Anterior Dystrophies
The primary therapy goal in patients with epithelial basement membrane or Bowman’s dystrophies is to maintain the integrity of the ocular surface.
A patient who is mildly symptomatic may be managed conservatively with lubrication. If symptoms worsen, medical treatments such as punctal plugs, hyperosmotic agents and topical or oral anti-inflammatories can be added. Although not a cure, patients can be fit in scleral lenses. These lenses completely vault over the cornea to protect the fragile epithelium while correcting vision compromised by irregular astigmatism.
The treatment of RCEs is tailored toward restoring the epithelium and decreasing patient pain, so placement of a bandage contact lens or amniotic membrane on the cornea is common. For any patient with an epithelial defect wearing a bandage lens, add a topical antibiotic for infection prophylaxis. Topical and/or oral anti-inflammatories can decrease matrix-metalloproteinase-9 (MMP-9) as well as speed up healing and prevent recurrent erosions. In a small study of seven patients with traumatic RCEs who were treated with a combination of topical steroid and oral doxycycline, all patients’ epithelial defects healed in 10 days or less with no recurrence of erosion for at least three months.5
All of these treatments support the stability of the ocular surface, but they won’t cure the condition. A more permanent approach is surgical intervention. Superficial keratectomy, where the epithelium is debrided with or without excimer laser phototherapeutic keratectomy (PTK), may be considered for superficial abnormalities of the cornea. These can be used to polish corneal irregularities or opacities within the anterior 10% to 20% of the cornea.6
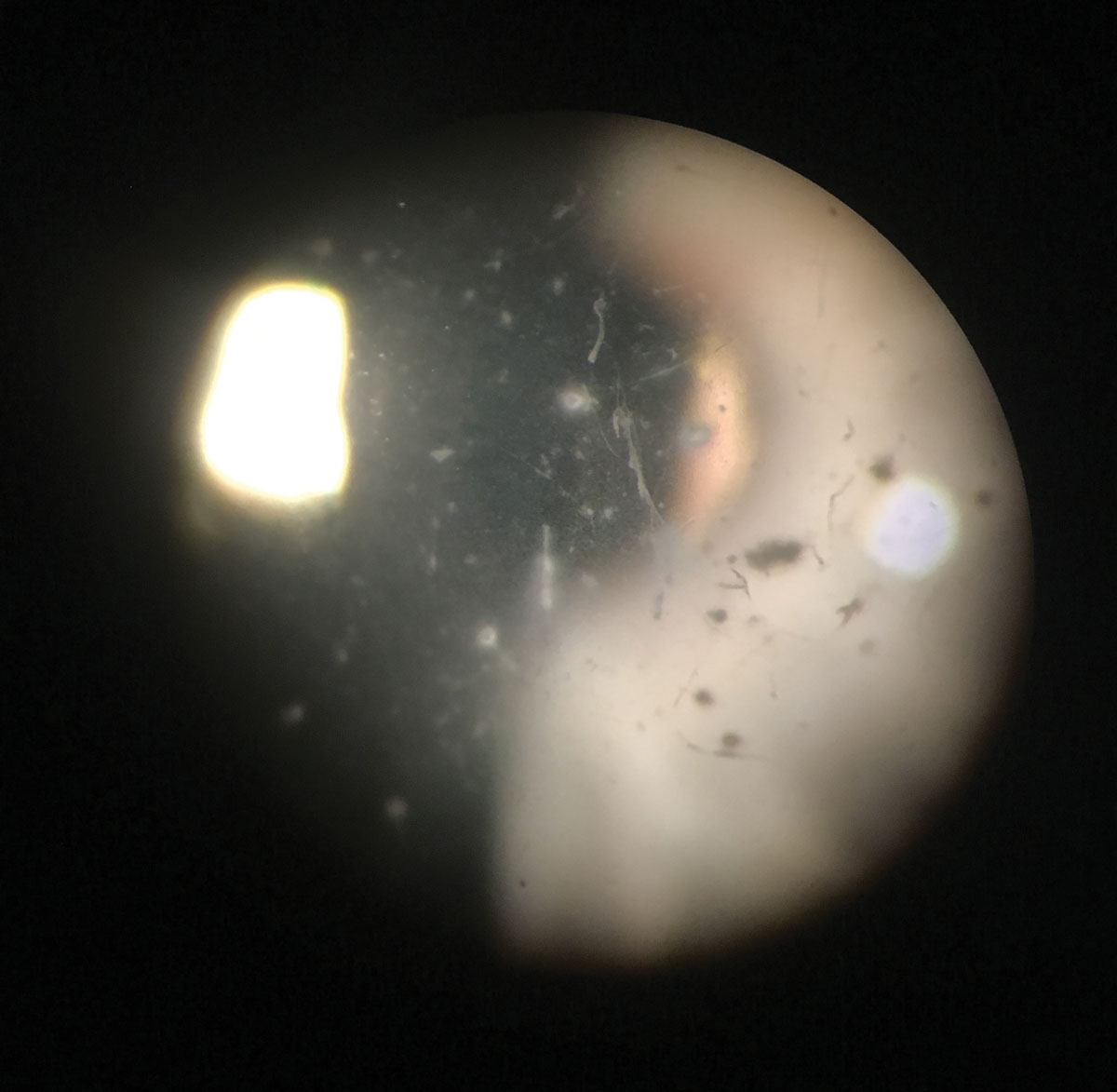 |
| Granular dystrophy type 2 (Avellino dystrophy) showing a central combination of line and circular opacities. Click image to enlarge. |
Stromal Dystrophies
The stroma represents the thickest layer of the cornea, measuring about 450μm. It is made up of dense, regular connective tissue that contains keratocytes, collagen, ground substance and water. Stromal dystrophies reside deep within the cornea and are typically treated in a conservative manner; but if stromal opacification presents, treatment options are limited.
Multiple stromal dystrophies can affect the cornea, but the most common are macular dystrophy, granular dystrophy, lattice dystrophy, Avellino dystrophy and Schnyder dystrophy.
Macular corneal dystrophy (MCD). This is the least common yet the most visually impairing stromal dystrophy. Despite its classification, MCD can also affect Descemet’s membrane and the endothelium. Caused by mutations in the CHST6 gene, MCD is the only autosomal recessive stromal dystrophy and is associated with an aggregation of glycosaminoglycans and mucopolysaccharides within the stroma.4
In affected patients, the cornea demonstrates bilateral corneal opacities that extend throughout the thickness of the stroma. A key slit-lamp differentiator for MCD is that the stromal opacities commonly extend out to the limbus. As the condition progresses, the opacities coalesce and guttata can develop within the endothelium, both leading to a poor visual outcome within the first three decades of life. These patients are commonly diagnosed at an early age and can experience attacks of irritation and photophobia. Further testing will reveal a slightly thinner than average stroma.
Therapeutic options are limited to tinted contact lenses and lubrication, which help reduce symptoms only to a certain extent. The sole treatment is penetrating keratoplasty (PKP), which is a full-thickness corneal transplant that replaces the damaged tissue. Lamellar keratoplasty is typically not indicated due to its high rate of disease recurrence because it involves removing only the anterior cornea and may leave a damaged endothelium attached to the graft.4
Granular corneal dystrophy. This condition is characterized by bilateral amorphous hyaline deposits that present within the first decade of life. It is an autosomal dominant dystrophy that has a mutation in the TGFβI gene.4 Symptoms include glare and photophobia, with decreased vision as it progresses. Patients normally do not experience vision loss until later in life. The cornea will show small breadcrumb-like hyaline deposits in the central superficial stroma that spare the limbus. In the later phase, these deposits increase in number, enlarge and extend deeper into the stroma. RCEs may also occur. Treatment options include PKP or deep anterior lamellar keratoplasty.
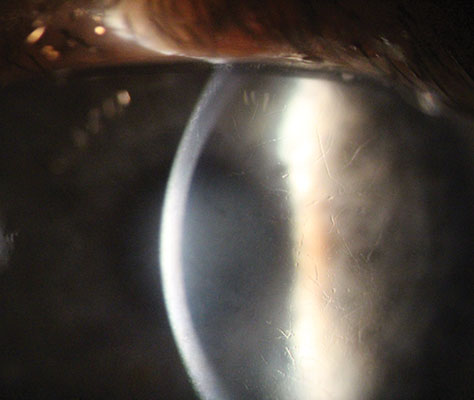 |
| Lattice dystrophy on high magnification. Note the progressive corneal haze. Click image to enlarge. |
Lattice corneal dystrophy. Caused by a mutation in TGFβI gene, this dystrophy manifests as amyloid deposits located in the anterior stroma.2 The deposits appear as refractile lines centrally in the anterior stroma, while the peripheral cornea remains largely unaffected. Later, these refractile lines progress into stromal opacifications, which can cause patients to be symptomatic with blurred vision. Some patients may lack the typical appearance of the stromal disease and only experience symptoms from RCEs.2
Initially, management is focused on treating any RCEs that occur; but if the opacities become visually significant, PKP may be indicated. Unfortunately, there is a high rate of recurrence of lattice corneal dystrophy within the graft.4 For surface irregularities, PTK is indicated. Opacities deep within the stroma cannot be treated by PTK because of the depth of involvement. Patients risk corneal haze and overall decreased vision the deeper the ablation depth.
Avellino corneal dystrophy. Also known as granular corneal dystrophy type 2, Avellino is a combination of granular and lattice corneal dystrophies that is caused by a mutation in the TGFβI gene.2 The affected cornea presents with white-gray granular deposits within the anterior stroma, while the mid-posterior stroma has lattice-like lesions that move more centrally with age. In the late phase, an overall haze can present throughout the cornea.
Management includes copious lubrication, anti-inflammatories or PKP.
Schnyder corneal dystrophy. This normally presents within the first decade of life, with multiple fine polychromatic crystal deposits that tend to form in a ring shape near the limbus. These deposits are caused by an accumulation of cholesterol and phospholipids that have a strong association with systemic hypercholesterolemia.7 Patients under the age of 40 with this presentation should have their fasting serum cholesterol and triglyceride levels measured.
Schnyder corneal dystrophy is caused by a mutation in the UBIAD1 gene and is rare.4 Surgical intervention is seldom needed, as vision is typically unaffected until later in life when the opacities become more central. If vision is significantly affected, a corneal graft may be indicated.2
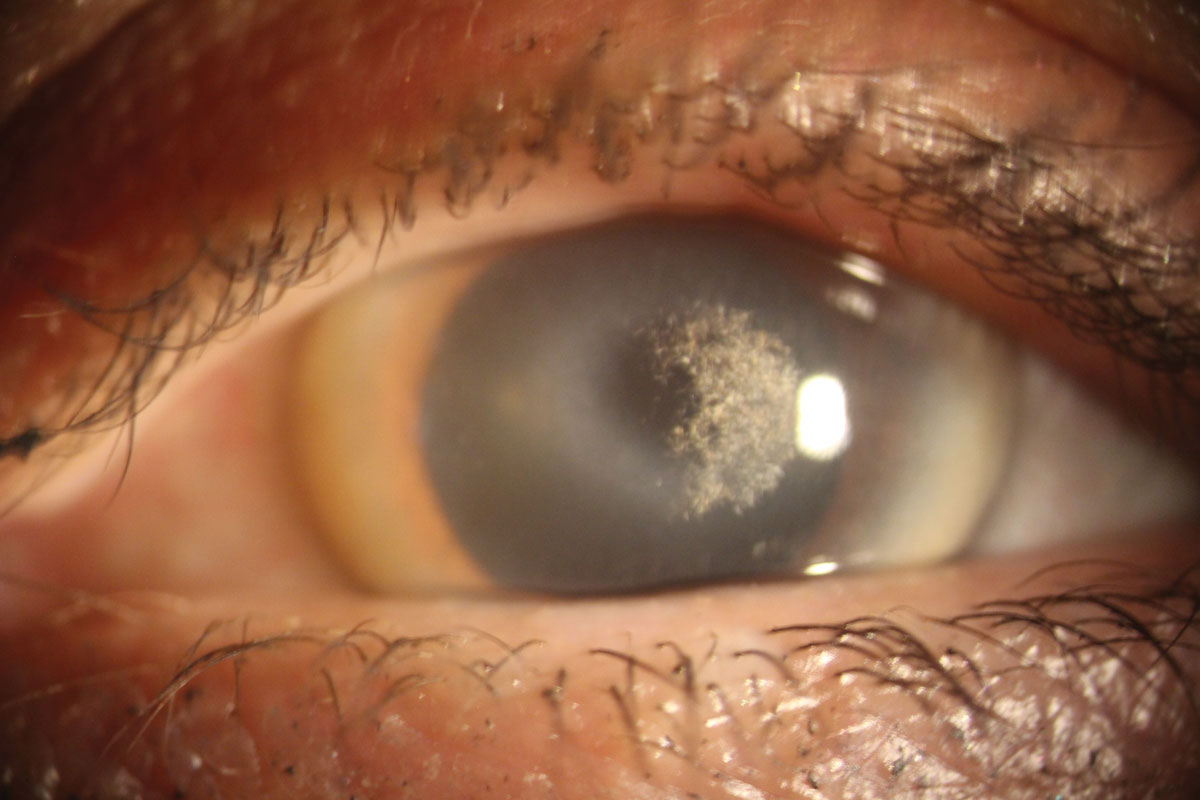 |
| Schnyder corneal dystrophy with an overlying scleral contact lens. Click image to enlarge. |
Endothelial Dystrophies
The endothelial cell layer represents the most posterior corneal layer. The endothelium is approximately 5μm thick and borders on its basement membrane, Descemet’s. This boundary layer’s main function is to regulate corneal hydration with active fluid transport to maintain a mostly dehydrated stroma.
Three main corneal dystrophies affect the posterior cornea: Fuchs’ endothelial dystrophy (FECD), posterior polymorphous dystrophy (PPCD) and congenital hereditary endothelial dystrophy (CHED). In all three, the endothelial pump function is disrupted, leading to an imbalance of hydration and resultant corneal edema.
Fuchs’ endothelial corneal dystrophy. This is the most common of all the corneal dystrophies in the United States with a prevalence of approximately 4% of the population over age 40.1 Fuchs’ dystrophy is inherited in an autosomal dominant pattern, affecting women more than men.
In FECD patients, the endothelial “pump” cells atrophy, leaving a fewer number of healthy pumping cells and corneal guttata (areas devoid of endothelial cells with thickened Descemet’s). Two hallmark endothelial cell morphology changes in FECD are pleomorphism (irregular change to cell shape) and polymegathism (irregular cell growth). As the endothelial cells reduce in number and pump ability continues to worsen, the corneal fluid equilibrium is disrupted and corneal edema ensues. Corneal edema results in loss of visual acuity and visual quality for patients where glare and haloes become a frequently cited visual disruption. In late stage FECD with chronic corneal edema, patients may develop painful bullae (blisters), which over time can alter the anterior corneal shape and epithelial integrity.
 |
| Posterior polymorphous dystrophy in a 17-year-old male. Click image to enlarge. |
Posterior polymorphous corneal dystrophy. This autosomal dominant corneal dystrophy results in abnormal vesicles with deep gray corneal haze at the level of Descemet’s and endothelium. The pathophysiology is thought to occur during gestation, and its ocular signs most commonly manifest in early childhood. In the corneal development of patients with PPCD, the endothelial layer is lined with variable amounts of epithelium-like squamous cells.4 The additional cell layers cause band-like adhesions and an abnormally thickened Descemet’s membrane. The abnormal cell bands can extend from the cornea and adhere to the iris, leading to peripheral anterior synechiae, putting PPCD patients at risk for glaucoma.4
Corneal edema typically isn’t progressive in patients with PPCD, but if vision in late childhood and early adulthood worsens, clinicians should initiate ancillary tests and a treatment plan to reduce edema.
Congenital hereditary endothelial dystrophy (CHED). The least common of the endothelial dystrophies, CHED has two distinctly recognizable forms: CHED1 and CHED2.4 A distinguishing factor of both CHED1 and CHED2 is bilateral thickened corneas with a ground glass appearance present at birth or infancy, which is different in onset from other endothelial dystrophies.4 CHED1 (autosomal dominant) patients manifest progressive corneal edema, tearing and photophobia, but do not have nystagmus.4 CHED2 (autosomal recessive) tends to be non-progressive, but presents with nystagmus plus occasional deafness (corneal dystrophy-perceptive deafness, a.k.a., Harboyan syndrome).4
 |
| Fuch’s dystrophy with early corneal edema. Note the classic orange peel appearance on the endothelium. |
Surgical Options
If any of these endothelial dystrophies result in progressive visual loss, relieving stromal edema is the most common treatment approach. Hyperosmotics may provide short-term relief in select patients, but for patients with progressing dystrophies, the best long-term treatment is a surgical approach.
For CHED1 and CHED2, due to stromal ground glass opacification, PKP is the surgical option of choice because corneal edema is less of a concern.4
In PPCD and FECD, in which surgical intervention is more dictated by corneal edema, an endothelial keratoplasty is preferred over a PKP. Endothelial keratoplasties are a better option due to less induced astigmatism, avoidance of incremental suture removal, lowered risk of rejection and better visual acuity potential compared with PKP.8 Descemet’s stripping endothelial keratoplasty and Descemet’s membrane endothelial keratoplasty (DMEK) are the two most commonly performed endothelial keratoplasties in the United States. Although both provide superior visual acuity results to PKP, DMEK has better visual acuity potential with a quicker visual recovery.9,10
Optometry plays a significant role for patients with corneal dystrophies because the initial diagnosis and referral, when necessary, is often made by an OD.
The optometrist’s fundamental task for the diagnosis and management of corneal dystrophies is to first determine which corneal layer is affected. Along with the case history, regular corneal topographies and corneal imaging can help to monitor for change and aid in the decision-making for management. Keep in mind that anterior dystrophies most commonly lead to more RCEs and opacification, while posterior dystrophies are associated with more progressive corneal edema. This tenet dictates medical and surgical treatment approaches.
When keratoplasty is necessary, anterior dystrophies require a full-thickness or anterior lamellar transplant; but for patients with posterior dystrophies, an endothelial keratoplasty should be the first choice.
Corneal dystrophies are rare, but correctly diagnosing these conditions changes the eye care landscape for a whole family.
Dr. Ibach is a residency-trained optometrist at Vance Thompson Vision in Sioux Falls, SD. He specializes in anterior segment surgical care, including cataracts, corneal diseases, glaucoma and refractive surgeries.
Dr. Zimprich is the current optometry resident at Vance Thompson Vision, where she will specialize in ocular disease as well as comanagement of refractive, cataract, corneal and glaucoma surgeries. She recently graduated from Massachusetts College of Pharmacy and Health Sciences.
1. Musch DC, Niziol LM, Stein JD, Kamyar RM, Sugar A. Prevalence of corneal dystrophies in the United States: estimates from claims data. Invest Ophthalmol Vis Sci. 2011 Sep 1;52(9):6959-63. 2. Krachmer J, Mannis M, Holland E (eds.). Cornea. 2nd ed, Vol 1. Philadelphia, PA: Elsevier; 2005: 898-902. 3. Jager RD, Lamkin JC. The Massachusetts Eye and Ear Infirmary Review Manual for Ophthalmology. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2005. 4. Klintworth GK. Corneal dystrophies. Orphanet J Rare Dis. 2009 Feb 23;4:7. 5. Dursun D, Kim MC, Solomon A, Pflugfelder SC. Treatment of recalcitrant recurrent corneal erosions with inhibitors of matrix metalloproteinase-9, doxycycline and corticosteroids. Am J Ophthalmol. 2001 Jul;132(1):8-13. 6. Paparo LG, Rapuano CJ, Raber IM, et al. Phototherapeutic keratectomy for Schnyder’s crystalline corneal dystrophy. Cornea. 2000 May;19(3):343-7. 7. Bagheri N, Wajda B (eds.). The Wills Eye Manual. 7th ed. Philadelphia: Wolters Kluwer; 2017. 8. Lee WB, Jacobs DS, Musch DC, et al. Descemet’s stripping endothelial keratoplasty: safety and outcomes: a report by the American Academy of Ophthalmology. Ophthalmology. 2009 Sep;116(9):1818-30. 9. Tourtas T, Laaser K, Bachmann BO, et al. Descemet membrane endothelial keratoplasty versus Descemet stripping automated endothelial keratoplasty. Am J Ophthalmol. 2012 Jun;153(6):1082-90. 10. Hamzaoglu EC, Straiko MD, Mayko ZM, et al. The first 100 eyes of standardized Descemet stripping automated endothelial keratoplasty versus standardized Descemet membrane endothelial keratoplasty. Ophthalmology. 2015 Nov;122(11):2193-9. |