Corneal Dystrophies Front to Back
This stepwise approach can help you diagnose and manage these conditions.
By Stephanie Fromstein, OD
 |
Release Date: September 15, 2020
Expiration Date: September 15, 2023
Estimated Time to Complete Activity: 2 hours
Jointly provided by Postgraduate Institute for Medicine (PIM) and Review Education Group
Educational Objectives: After completing this activity, the participant should be better able to:
- Describe the commonly confused corneal dystrophies.
- Use the clinical exam and diagnostic technology to distinguish different corneal dystrophies.
- Review the prevalence and genetic patterns of corneal dystrophies.
- Comprehend the implications for patients and their families.
- Discuss the various treatment options.
Target Audience: This activity is intended for optometrists engaged in the care of patients with corneal dystrophies.
Accreditation Statement: In support of improving patient care, this activity has been planned and implemented by the Postgraduate Institute for Medicine and Review Education Group. Postgraduate Institute for Medicine is jointly accredited by the Accreditation Council for Continuing Medical Education, the Accreditation Council for Pharmacy Education, and the American Nurses Credentialing Center, to provide continuing education for the healthcare team. Postgraduate Institute for Medicine is accredited by COPE to provide continuing education to optometrists.
Faculty/Editorial Board: Stephanie Fromstein, OD, Illinois College of Optometry.
Credit Statement: This course is COPE approved for 2 hours of CE credit. Course ID is 69301-AS. Check with your local state licensing board to see if this counts toward your CE requirement for relicensure.
Disclosure Statements:
Dr. Fromstein has nothing to disclose.
Managers and Editorial Staff: The PIM planners and managers have nothing to disclose. The Review Education Group planners, managers and editorial staff have nothing to disclose.
Corneal dystrophies are some of the most fascinating—and frustrating—corneal findings eye care providers encounter. Along with anomalies, degenerations, inflammations and infections, it can be challenging to navigate appropriate history and clinical findings to narrow down an accurate diagnosis.
Dystrophies do, however, have several distinguishing features that can help in their identification. Owing to their genetic basis, dystrophies are most often bilateral and relatively symmetric.1-3 They are usually noted in patients younger than age 20 and tend to progress slowly over time; generally, there is no acute change in vision or symptoms, but rather a gentle decline into relative dysfunction.1-3 The vast majority of corneal dystrophies are not only hereditary but also autosomal dominant, meaning that immediate family members will share clinical findings, which may aid in diagnosis.
On clinical exam, associated corneal findings tend to present with no other related ocular or systemic disease. Perhaps most importantly for diagnosis, these conditions are located in a single layer of the cornea; thus, accurate determination of location can go a long way toward narrowing the field of potential diagnostic contenders.1-3
Corneal degenerations, on the other hand, represent a slow and steady deterioration of corneal tissue with wear and tear. Degenerations usually present as unilateral/asymmetric changes in older patients, often with no underlying genetic etiology. Corneal changes tend to present predominantly peripherally and may impact several corneal layers. These differences from dystrophic changes can be essential in helping to differentiate the two types of corneal changes.
Epithelial Dystrophies
Moving front to back, we start with dystrophies localized to the corneal epithelium, a five-layer thick structure bound on the southern border by a basement membrane. There are only two epithelial dystrophies, significantly narrowing the field for an accurate diagnosis. Moreover, one of the epithelial dystrophies is strikingly common, while the other is exceedingly rare.
Epithelial basement membrane dystrophy (EBMD). When discussing prevalence, EBMD is the most common dystrophy, with a prevalence of more than 40% in the general population. In fact, this condition is so prevalent—jumping to 70% in those over 50 years old—that there is debate within the eye care community as to whether it may instead represent a corneal degeneration. A lack of clear inheritance pattern also speaks to this suspicion.4
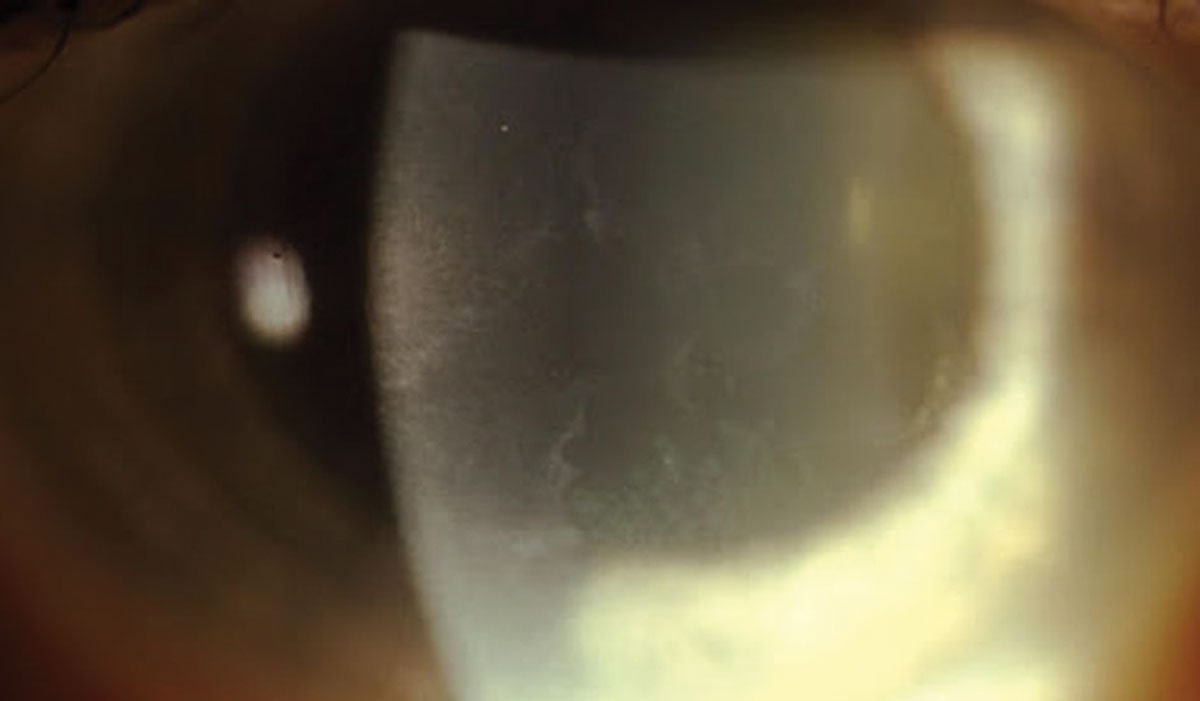 |
| Fig. 1. Large “maps” of redundant epithelium in EBMD. Click image to enlarge. |
This condition—by any definition and any other name (anterior basement membrane dystrophy, map-dot dystrophy, microcystic epithelial dystrophy and Cogan’s microcystic dystrophy)—represents a pathognomonic set of superficial corneal findings due to a faulty epithelial basement membrane.1 The overzealous basement membrane extends abnormally into the epithelium, heaping up the epithelial tissue and forming large well-demarcated areas of opacification known as maps (Figure 1). Migrating cellular material becomes trapped and manifests as corneal dots and other unnamed cystic changes. Adjacent rows of thickened and elevated epithelium appear as fingerprints (Figure 2).1-3
The clinical presentation of these findings can range from evident to elusive; where the etiology of symptoms is opaque, sodium fluorescein (NaFl) can be an invaluable diagnostic aid, as the heaped-up epithelial tissue—devoid of fluorescein—appears black, highlighted by the pooling of green around it, in a configuration known as negative staining.5 (As a side note, it is due to this very finding that I stress to my students to record “clear to NaFl” on healthy corneal evaluations rather than “(-)NaFl,” as this insinuates that the patient has been, or should be, evaluated for EBMD).
When accurately identified, EBMD is noted more often in female patients and increasingly with advancing age.4 Cases are largely asymptomatic or mild, with the most common symptoms relating to visual changes—usually blur (worse in the morning) or diplopia. Non-specific symptoms such as foreign body sensation and dry eye may also be present. Most often these cases subsist with simple monitoring until complications arise in the form of recurrent corneal erosion (RCE).6
The thickened and redundant basement membrane means that epithelial hemidesmosomes are not able to do their job of anchoring the epithelium into the anterior stroma. This inability of the epithelium to “anchor down” explains the high association of EBMD with RCE. It is estimated that about 33% of EBMD patients will experience RCE at some point in their lives, making it the second most common underlying cause behind trauma.7
As a clinical note, the vast majority of EBMD-mediated RCE is found in the inferior one-third of the cornea, so be sure to direct special attention to this area during your slit-lamp evaluation.8
If the patient is symptomatic for visual changes, foreign body sensation or dry eye, firstline treatment is lubrication. For RCE prevention, hypertonic salt solutions are a mainstay of therapy to decrease corneal swelling and keep the epithelial tissue anchored to the underlying stroma.9 Topical therapy, such as steroids, and oral therapy, such as doxycycline (by way of metalloproteinase-9 suppression), are also effective therapies for preventing RCE.10
 |
| Fig. 2. Adjacent rows of thickened and elevated epithelium (“fingerprints”) viewed with NaFl in a patient with EBMD. Click image to enlarge. |
Moderate to severe cases—most commonly those with a positive history of RCE—may require surgical intervention to remove the irregular epithelium. This debridement can be performed with a round blade, spatula, diamond burr or excimer laser. Excimer-laser mediated phototherapeutic keratectomy (photorefractive keratectomy, but for the purpose of minimizing the irregular epithelium and scarring) is preferred by many surgeons for this indication.
Stromal micropuncture has also been used with good success to induce scar tissue to anchor the epithelium to the underlying tissue. Long-term management may additionally include soft or gas-permeable contact lenses to protect the cornea from further erosion and mask any residual irregularity. Lamellar keratoplasty is generally not indicated.9
Meesman corneal dystrophy. This epithelial dystrophy is a rare condition despite its autosomal dominant inheritance pattern. Also known as juvenile hereditary epithelial dystrophy, the condition manifests early in life with intraepithelial cysts and vesicles due to localized keratin dysfunction. These cysts are generally small and filled with granular, filamentary material known as “peculiar substance.” While they may extend limbus-to-limbus, they tend to be more concentrated centrally and interpalpebrally, and are best viewed via retroillumination.
The condition is largely asymptomatic other than mild visual changes (blur, glare, light sensitivity) and discomfort if and when the cysts rupture onto the epithelial surface.1-3 If symptomatic, lubricants and debridement may be considered as well as lamellar keratoplasty in rare cases where significant opacification has led to decreased acuity.11
Bowman’s/Anterior Stromal Dystrophies
As we move deeper into the cornea, we arrive at Bowman’s layer, which is notable because it is much more likely to scar than its epithelial counterpart.12 Much like with the epithelial dystrophies, there are only a handful of dystrophies to consider, once accurately localized.
While it is nearly impossible to identify changes specific to Bowman’s layer on slit-lamp examination, a few clinical pearls can aid in differentiation. Anteriorly, the appearance of the dystrophic changes in Bowman’s layer do not resemble those of the epithelium; thus, these are not easily conflated.
Posteriorly, when compared with dystrophies of the anterior stroma, it is helpful to recall corneal anatomy; Bowman’s layer dystrophies will appear subepithelial and deposition will appear to be in a very thin layer just below the epithelium. Anterior stromal dystrophies present deeper than Bowman’s layer dystrophies, and the deposits are dispersed more diffusely throughout a thicker slice of the corneal landscape.
As such, Bowman’s layer dystrophies are not quite epithelial while not quite stromal, which can be diagnostic unto itself. Anterior segment OCT can also be helpful in isolating the deposits, should this technology be available in-office.
Until recently, these dystrophies were considered one and the same, and even since being distinguished (based on electron microscopy and not on clinical findings), they remain variations rather than completely distinct entities. Given their similitude, the three Bowman’s layer dystrophies (Reis-Buckler’s, Thiel-Behnke and Grayson-Willbrandt/Stocker-Holt) can be considered as a single clinical entity.13
All Bowman’s layer dystrophies are bilateral, symmetric and autosomal dominant. Clinical examination reveals central ring-shaped subepithelial opacities that generally appear in early childhood. Compared with epithelial dystrophies, these patients are more likely to be symptomatic, complaining of visual changes (blur, photophobia, diplopia) as well as foreign body sensation and pain, often due to RCE.1-3
If symptoms are mild, they can often be managed with lubrication. More severe cases may require debridement and/or keratoplasty for those with significant scarring impacting acuity. RCE should be managed as indicated.1-3
Stromal Dystrophies
The stroma takes up the bulk of corneal real estate (90%) and, accordingly, many of the dystrophies demonstrate clinical changes in this layer.14 There are a larger number of stromal dystrophies compared with any other layer, though frequent recategorization means that the actual number varies slightly as more advanced technology allows us to more accurately localize our findings. While there is massive variation among the stromal dystrophies, they can broadly be narrowed based on prevalence, type of deposition and location within the layer (anterior or posterior).
Lattice dystrophy. This is the most prevalent stromal dystrophy, displaying autosomal dominant inheritance and an early onset of symptoms (usually within the first decade). The condition is characterized by fine, branching irregular amyloid deposits arranged in fine lines in the anterior stroma.1-3 These deposits may initially be surrounded by haze that fibroses over time, causing the refractile deposits to become opacified.
There were formerly two types of lattice dystrophy, distinguished mainly by the presence of systemic manifestations of amyloid accumulation (Type I has no systemic associations, while Type II has been associated with connective tissue disorders and ataxia). However, Type II is now considered a systemic disorder with ocular findings, not a separate corneal dystrophy.1-3,15
Decreased acuity is likely a result of the deposition and attendant haze. Reduced corneal sensitivity is common.1-3,15 RCE may also occur as a result of disruption of the hemidesmosomes anchoring the epithelium to the stroma, though this time the interruption comes from below.
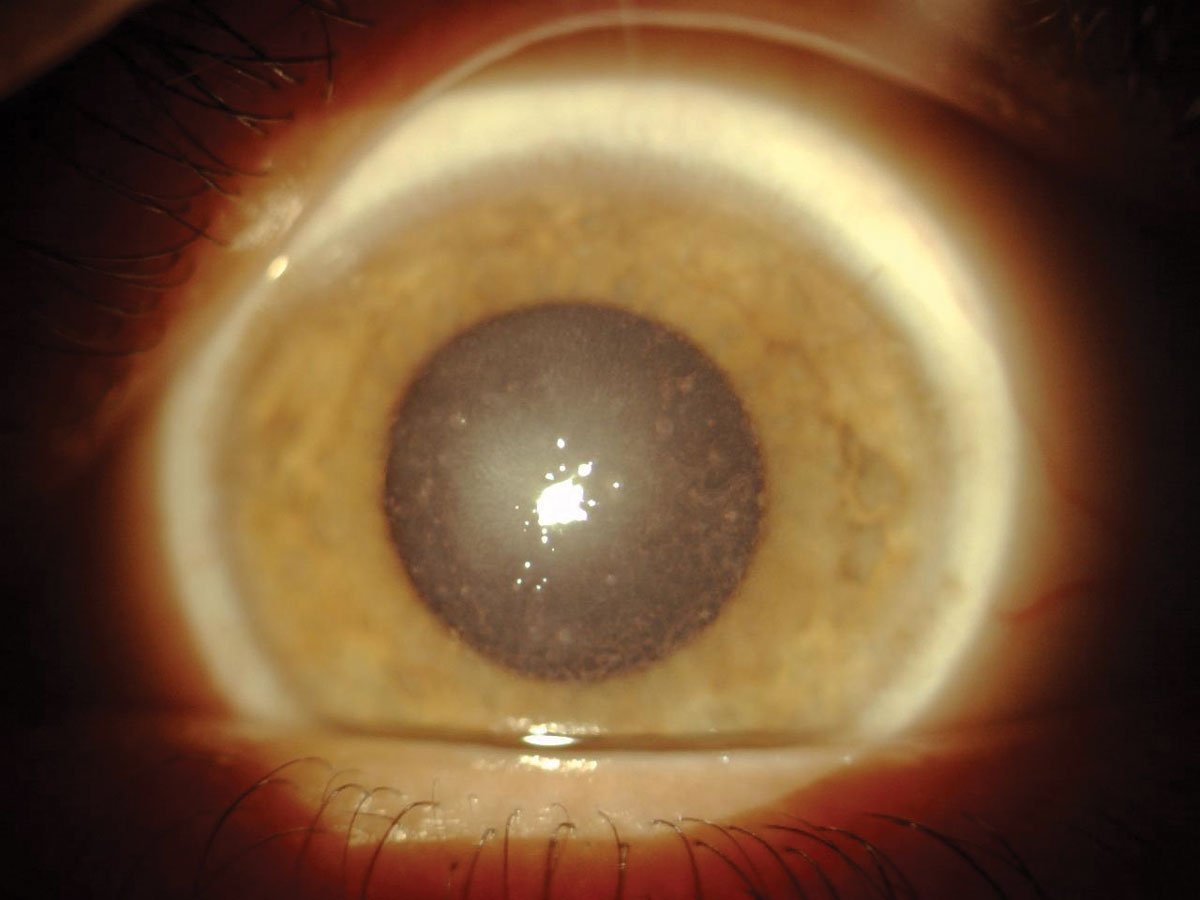 |
| Fig. 3. Stromal hyaline deposits seen in a patient with granular dystrophy. Click image to enlarge. |
Granular dystrophy. This condition is next in line in prevalence, with two distinct iterations each demonstrating autosomal dominant inheritance and an onset in the first or second decade of life. Granular dystrophy Type I (classic type) is characterized by small, discrete white granules composed of hyaline in the anterior stroma (Figure 3).1-3 These demarcated deposits are initially separated by clear spaces, but as the condition progresses, the breadcrumb/snowflake-like deposits become more confluent.
Symptoms are initially mild but progressive, and patients are likely to suffer from consequent decreased acuity requiring treatment.1-3 This dystrophy may cause decreased acuity, decreased corneal sensitivity and RCE. In contrast to other corneal dystrophies, lattice dystrophy is more likely to be unilateral or largely asymmetric.1-3
The second form of granular dystrophy (Type II)—also known as granular-lattice or Avellino dystrophy—combines features of both granular and lattice dystrophy. Anterior stromal, discrete gray-white opacities and lattice lesions in the mid- and posterior stroma are pathognomonic for the condition.16
In contrast to classic lattice dystrophy, the branches do not cross and form the classic “lattice” pattern for which the dystrophy is named, in addition to other more subtle differentiating features.15 The condition tends to have more severe symptoms with an earlier onset compared with its classic counterpart, owing in part to its autosomal dominant nature with high penetrance.1-3
Macular dystrophy. Rounding out the top three stromal dystrophies is macular dystrophy, which is unique in several ways. First, it is the only autosomal recessive stromal dystrophy, meaning that the patient may not have the marked family history that will be noted in previous conditions. Despite this, it still shows a relatively high prevalence, even greater than lattice and granular in some studies. It also has a tendency to extend limbus-to-limbus (not sparing the periphery) and anterior-to-posterior stroma.1-3
Clinical examination reveals gray-white, ill-defined focal opacities composed of glycosaminoglycan. Diffuse cloudiness results from changes in the collagen fibril arrangement. Interestingly, as the regular collagen tissue is replaced by fibrotic tissue, the condition may be associated with corneal thinning.17 Given that it breaks so many of the rules of corneal dystrophies as well as its similar appearance to a host of other corneal findings (including inflammatory corneal signs seen as a result of infection or other corneal insult), this can be a challenging identification. However, symptoms of visual changes, decreased corneal sensitivity and the potential for RCE remain consistent with other stromal dystrophies.
Fleck dystrophy. Beyond these ‘big three,’ there are several increasingly rare stromal dystrophies that bear mentioning for completeness. Fleck or speckled dystrophy is autosomal dominant with onset in the first decade. It may be unilateral or relatively asymmetric. The condition is most often mild, with patients reporting no symptoms or occasional photophobia.
On clinical examination, very small gray-white opacities can be seen scattered in the stroma, giving the appearance of dandruff. While not entirely clear why, there is an association with other ocular findings, including keratoconus, ocular dermoids, cataracts and papillitis, which may speak to a defect of collagen metabolism, which has not yet been fully elucidated.1-3
Central crystalline dystrophy of Schnyder. This autosomal dominant condition presents in the first decade of life with central disciform opacities often surrounded by a finding of dense arcus (related but distinct). This combination of central and peripheral findings is distinct and pathognomonic for this dystrophy.
Deposits consist of polychromatic needle-shaped crystals composed of cholesterol, reflecting a localized disorder of cholesterol metabolism. This localized dysfunction can be, and often is, exacerbated by systemically elevated serum cholesterol. Given that 50% have elevated levels of cholesterol, every patient presenting with Schnyder dystrophy warrants a hyperlipidemia workup.1-3
Central cloudy dystrophy of Francois. This rare, autosomal dominant dystrophy is minimally symptomatic. Like Schnyder, it is also thought to be a result of cholesterol deposition, but with far less visual complication. The polygonal, cloudy gray opacities are thought to be an inherited form of Crocodile Shagreen, giving a leather-like appearance to the posterior stroma.1-3
Gelatinous drop-like dystrophy. This is one of the least common but most visually devastating corneal dystrophies. It is the result of a localized dysfunction of amyloid in the cornea that deposits subepithelially and stromally, leading to significant signs and symptoms. For this reason, it is sometimes classified alongside the more anterior dystrophies. Patients complain of severe redness, irritation, photophobia and tearing.
A clinical examination may reveal decreased acuity as well as centrally raised mounds of amyloid. The late-stage appearance of the condition is referred to as a “nubbly mulberry” surface, which almost invariably requires intervention.1-3
Therapeutic options. Treatment for stromal dystrophies depends on the severity of signs and symptoms. Mild forms may be monitored or may encourage use of lubrication for any discomfort or foreign body sensation. More severe cases may require phototherapeutic keratectomy or other forms of debridement, which can be effective in delaying the need for corneal transplantation by up to two years.17
If acuity and erosion continue to be problematic, keratoplasty (lamellar or otherwise) may be required for management. While penetrating keratoplasty (PKP) was formerly a mainstay of treatment, studies suggest deep anterior lamellar keratoplasty (DALK) may also be considered with similar acuity outcomes and a lower risk of complication.17
Keep in mind, the genetic coding of this information is not eliminated along with the dysfunctional tissue in the case of debridement and transplantation. Thus, recurrence (even in the donor tissue) ranges from possible to probable depending on the individual dystrophy and is more likely to occur with stromal dystrophies (most especially with lattice corneal dystrophy).18 The patient should be made aware of this likelihood before proceeding with surgery.
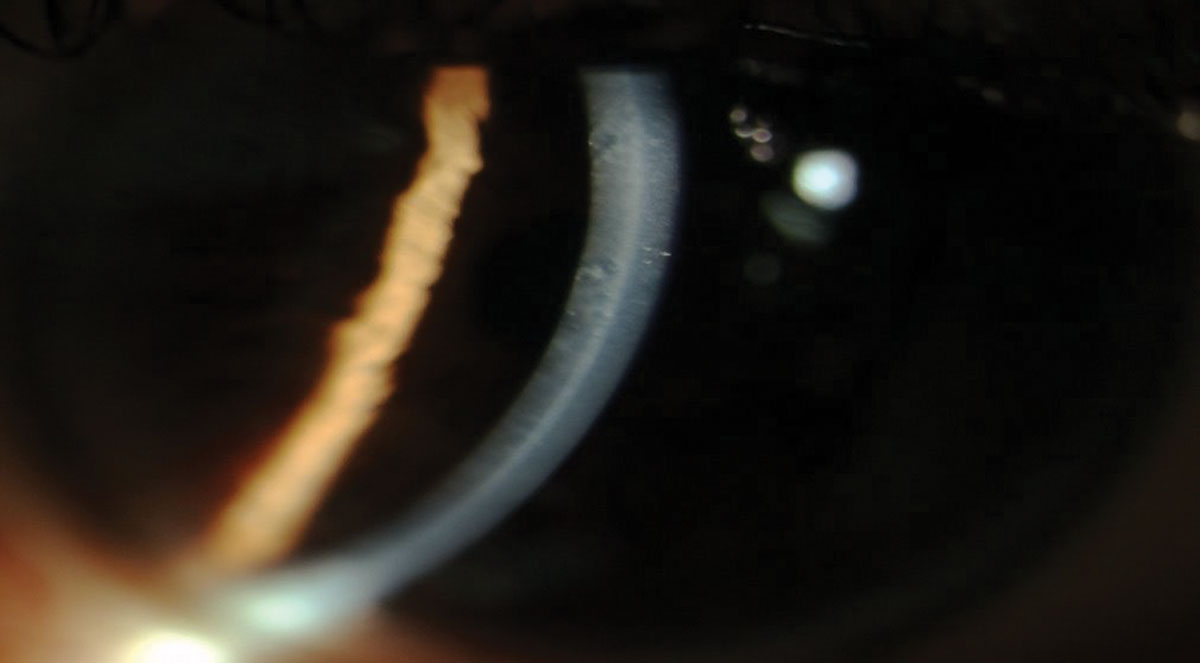 |
| Fig. 4. Endothelial vesicles in a patient with PPMD. Click image to enlarge. |
Endothelial Dystrophies
There are three endothelial dystrophies of note, two of which are significantly more common and much more likely to be diagnosed in your chair. Given the endothelium’s principal enterprise as the corneal dehumidifier, any disfunction of the endothelium can lead to areas of localized or diffuse corneal edema as well as the attendant complications of decreased acuity and epithelial microcysts.
Posterior polymorphous dystrophy (PPMD). This autosomal dominant dystrophy can impact both Descemet’s membrane as well as the endothelium. PPMD can have a wide variety of presentations from linear, parallel, bandlike configurations to vesicles to irregular configurations with scalloped edges, mimicking a break in Descemet’s membrane (Figures 4 and 5).1-3 While the presentation varies widely, it tends to be consistent among families, so examination of an immediate family member can give an indication of what type of findings you are likely to see in your own patient.19
PPMD is, in a sense, the EBMD of the endothelium, with an abnormal proliferation of cells leading to a wide variety of clinical findings. Essentially, the endothelial cells begin to act as epithelial cells and proliferate, causing the accumulation of an abnormal basement membrane and the thickening of Descemet’s membrane.19 This abnormal proliferation can have a significant impact on endothelial function and may cause abnormalities in adjacent structures (most commonly the iris and angle structures), leading to increased incidence of secondary glaucoma.1-3,19
If PPMD is suspected, gonioscopy should be performed to evaluate the angle structures.19 Iridocorneal endothelial and mesodermal dysgenesis syndromes may present similarly and should be considered as differential diagnoses; a strong family history can help you make an accurate diagnosis.
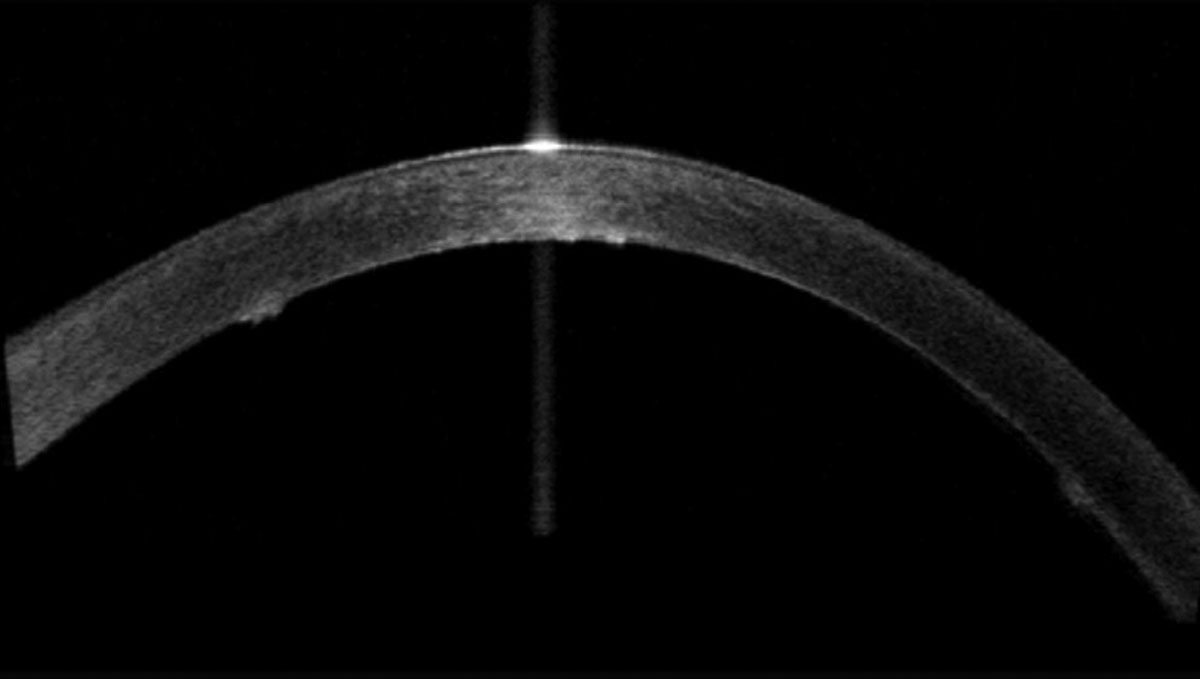 |
| Fig. 5. Endothelial vesicles as viewed with anterior segment OCT on a patient with PPMD. Click image to enlarge. |
Fuchs’ endothelial dystrophy. This common endothelial dystrophy is marked by bilateral, accelerated endothelial cell loss. It is autosomal dominant and more common in women, usually with a symptomatic onset later in life (60s and 70s).1-3,20 It is slowly progressive over time but may be acutely accelerated by cataract surgery, given the demographics of affected individuals. Early on, patients may complain of photosensitivity or decreased acuity. Early clinical signs are dominated by corneal guttata—seen as tiny dark spots on specular reflection. Pigment may also be noted on the dysfunctional endothelium.1-3
While seemingly a void, these dark spots represent not an absence of tissue but rather a function of the tissue being pushed out of the plane of focus. This happens due to a buildup of basal lamina (collagen), causing Descemet’s membrane and the endothelium to bulge posteriorly in tandem into the anterior chamber (and out of the plane of focus).1-3
To avoid total corneal decompensation, the endothelium remodels to help preserve whatever functionality it can. It undergoes a decrease in the number of endothelial cells, causing the remaining cells to both change shape (pleomorphism) and increase in size (polymegathism) as the functional endothelial cells try to cover the workload of the dysfunctional ones. These can both be monitored with specular microscopy, where available.1-3,20
As the condition progresses, patients are likely to experience decreased acuity secondary to profound corneal edema. This can and should be monitored by serial pachymetry. Depending on the level of swelling, stromal edema or epithelial microcysts and/or bullae may also be noted.20
Congenital hereditary endothelial dystrophy (CHED). This endothelial dystrophy is quite rare and most often diagnosed at birth. The patient will present with bilateral, limbus-to-limbus diffuse corneal edema secondary to the dysfunctional endothelium. This is referred to as a “ground glass” appearance.
There are both autosomal dominant and autosomal recessive iterations, with varying degrees of associated systemic findings including nystagmus and hearing loss. The condition may be stationary or slowly progressive.1-3
Management techniques. Treatment for endothelial dystrophies is mainly targeted at minimizing the secondary corneal edema. Mild cases may be monitored, often with serial pachymetry. Moderate conditions may be treated with hypertonic agents or even a hairdryer to minimize corneal hydration. Global decompensation often requires some form of corneal transplantation.
Fuchs’ dystrophy was once the chief reason for a full thickness corneal transplant; as our ability to transplant more selectively has improved, these patients can now be treated with a lamellar transplant such as Descemet’s membrane endothelial keratoplasty, Descemet’s stripping endothelial keratoplasty and Descemet’s stripping automated endothelial keratoplasty.20 This has been a revelation for patients in terms of recovery time and outcomes, and has anecdotally impacted the number of patients willing to pursue treatment for the condition.
There are a wide variety of clinical findings associated with corneal dystrophies. This variety—along with an accurate localization of the findings—makes diagnosis of these conditions relatively straightforward. In any given corneal layer, there are no more than three common contenders, and in no case do the various conditions within a layer resemble each other closely.
This means that armed with an entry-level understanding of the prevalence, clinical features and associated findings, diagnosing these conditions should be relatively effortless. Features shared within a given lamella, such as probable signs, symptoms and complications, further simplify and streamline management. While dystrophies on the aggregate can and often do seem intimidating, as with most things, a stepwise approach yields dividends in terms of diagnosis and treatment.
Dr. Fromstein is an assistant professor at the Illinois College of Optometry and practices at the Illinois Eye Institute in the Cornea and Contact Lens department in Chicago.
1. Kanski J, Bowling B. Clinical Ophthalmology A Systematic Approach. Elsevier Health Sciences; 2011. 2. Yanoff M, Duker J. Ophthalmology. 4th ed. Elsevier Health Sciences; 2014. 3. Rapuano C. Cornea: Color Atlas & Synopsis of Clinical Ophthalmology. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2012. 4. Werblin TP, Hirst LW, Stark WJ, et al. Prevalence of map-dot-fingerprint changes in the cornea. Br J Ophthalmol. 1981;65(6):401-9. 5. Ramsey A. Vital stains: what you really need to know. RCCL. April 2011. www.reviewofcontactlenses.com/article/vital-stains-what-you-really-need-to-know. Accessed August 18, 2020. 6. Reidy JJ, Paulus MP, Gona S. Recurrent erosions of the cornea: epidemiology and treatment. Cornea. 2000;19(6):767-71. 7. Miller DD, Hasan SA, Simmons NL, et al. Recurrent corneal erosion: a comprehensive review. Clin Ophthalmol. 2019;13:325-35. 8. Hykin PG, Foss AE, Pavesio C, et al. The natural history and management of recurrent corneal erosion: a prospective randomised trial. Eye(Lond). 1994;8(Pt 1):35-40. 9. Stephenson M. When and how to treat EBMD. Rev Ophthalmol. 2019;26(9):58-64. 10. Dursun D, Kim MC, Solomon A, et al. Treatment of recalcitrant recurrent corneal erosions with inhibitors of matrix metalloproteinase-9, doxycycline and corticosteroids. Am J Ophthalmol. 2001;132(1):8-13. 11. Greiner JV, Lindsay ME, Kenyon KR, et al. Meesmann epithelial corneal dystrophy: recurrence following photorefractive keratectomy. Can J Ophthalmol. 2017;52(6):e211-3. 12. Lu L, Reinach PS, Kao WW. Corneal epithelial wound healing. Exp Biol Med (Maywood). 2001;226(7):653-64. 13. Tanhehco TY, Eifrig DE, Schwab IR, et al. Two cases of Reis-Bucklers corneal dystrophy (granular corneal dystrophy type III) caused by spontaneous mutations in the TGFBI gene. Arch Ophthalmol. 2006;124(4):589-93. 14. Levin LA, Nilsson S, Ver Hoeve J, et al. Adler’s Physiology of the Eye. 11th ed. Elsevier Health Science; 2011. 15. Moshirfar M, West W, Ronquillo Y. Lattice corneal dystrophy. Treasure Island (FL): StatPearls; 2020. 16. Abazi Z, Magarasevic L, Grubisa I, et al. Individual phenotypic variances in a family with Avellino corneal dystrophy. BMC Ophthalmol. 2013;13:30. 17. Aggarwal S, Peck T, Golen J, et al. Macular corneal dystrophy: a review. Surv Ophthalmol. 2018;63(5):609-17. 18. Moshirfar M, Bennett P, Ronquillo Y. Corneal dystrophy. Treasure Island (FL): StatPearls; 2020. 19. Guier CP, Patel BC, Stokkermans TJ, et al. Posterior polymorphous corneal dystrophy. Treasure Island (FL): StatPearls; 2020. 20. Sarnicola C, Farooq AV, Colby K. Fuchs endothelial corneal dystrophy: update on pathogenesis and future directions. Eye Contact Lens. 2019;45(1):1-10. |